
Abstract
To link microbial community 16S structure to a measured function in a natural soil, we have scaled both DNA and β-glucosidase assays down to a volume of soil that may approach a unique microbial community. β-Glucosidase activity was assayed in 450 individual aggregates, which were then sorted into classes of high or low activities, from which groups of 10 or 11 aggregates were identified and grouped for DNA extraction and pyrosequencing. Tandem assays of ATP were conducted for each aggregate in order to normalize these small groups of aggregates for biomass size. In spite of there being no significant differences in the richness or diversity of the microbial communities associated with high β-glucosidase activities compared with the communities associated with low β-glucosidase communities, several analyses of variance clearly show that the communities of these two groups differ. The separation of these groups is partially driven by the differential abundances of members of the Chitinophagaceae family. It may be observed that functional differences in otherwise similar soil aggregates can be largely attributed to differences in resource availability, rather than to the presence or absence of particular taxonomic groups.
Similar content being viewed by others


Introduction
The composition of soil microbial biomass is overwhelmingly diverse, up to 107 different species in a gram of soil (Gans et al., 2005), and these remarkable assemblages mediate a vast number of ecosystem processes associated with nutrient cycling, C dynamics and soil health. Contemporary high-throughput sequencing techniques are rapidly increasing our knowledge of what organisms live in soil, and their phylogenetic diversity. However, directly linking this information, essentially ‘census’ data, with actual ecosystem processes is needed in order to understand how interacting microbial communities evolve and function (Morales and Holben, 2011). For natural microbial communities, we do not yet know the relationship between diversity and function, because it has not been analyzed at an appropriate spatial scale. Until recently, identifying such structure–function linkages in soil has been hindered by the amount of biomass needed for experiments; the high-throughput sequencing of soil metagenomes has been done on quantities of soil too large to consider as a defined habitat for a deconvolvable microbial community.
Unlike more well-mixed or constrained habitats, such as termite guts or microbial mats, the 3-dimensional nature of soil, presenting chemical and physical heterogeneities and barriers, makes it difficult to assign the microbes, reported in a metagenome, to particular ecological roles. Additionally, microbial census data, such as 16S sequences, are analyzed in the context of existing sequence databases, and soils are notoriously home to a wealth of uncultured or unclassified microorganisms (Dokic et al., 2010). Shotgun metagenomes seek to provide greater genetic information to yield functional inferences, but their usefulness is often hampered by the challenges associated with (1) de novo assembly of short read sequence data from a complex assemblage, (2) the availability of too few appropriate reference genomes to which metagenome fragments can be aligned and (3) poor accuracy of binning of the sequenced fragments into appropriate groups or potential operational taxonomic units (OTUs) that reflect species within the assemblage (Wooley et al., 2010).
Further confusing our ability to understand the microbial communities in natural systems, such as soil, is the fact that what the community is doing is likely far more complex than which species are present, based on the likely presence of functional redundancies within the community and metabolic versatility within single species. Unfortunately, classical soil enzyme assays, though simple to conduct, reveal the potential function of a sample, not the actual expressed function in situ. Efforts to study the in situ activities in the soil by directly analyzing mRNAs have proven difficult, given the ephemeral nature of mRNA and the abundance of nucleases in soil (Cai et al., 2006; Pietramellara et al., 2009). Additionally, both enzyme assays and the mRNA analyses are limited; most enzyme assays will probably be reflective of multiple genes and organisms, and mRNA analyses are dependent on a priori sequence knowledge (Bailey et al., 2010; McGrath et al., 2010; Wang et al., 2011). Finally, both analyses have traditionally been conducted on relatively large sample sizes, compared with the scale at which microorganisms interact as a community, the latter of which would better able researchers to draw correlations (that is, (Kumaresan et al., 2011)) between the community structure and function.
The main objective of our research was to link microbial community structure to a measured function in a natural soil, at a scale approaching a defined habitat. We used β-glucosidase activity as a target function; it is a likely rate-limiting step in cellulose decomposition (van Zyl et al., 2010), and we have successfully analyzed this enzyme sequentially with measurements of total soil ATP as a surrogate for microbial biomass (Bailey et al., 2012). These biochemical assays were performed on individual soil aggregates, naturally occurring ‘clumps’ of soil less than a millimeter in diameter, to constrain the microbial community to a scale more representative of a community capable of interacting. These aggregates were then grouped according to enzyme activity level and the DNA extracted; combining similarly active aggregates was necessary to recover sufficient DNA for pyrosequencing. In these grouped samples, we observed that although the diversities of the microbial communities were similar, there were statistically significant differences in community structure between the high and low β-glucosidase activity aggregate groups.
Materials and methods
Soils
Soil was collected from a grassland field at the United States Department of Agriculture Conservation Field, near Pullman, Washington (fine-silty, mixed, superactive, mesic Pachic Ultic Haploxeroll). Macroaggregates ranging in size from 841 to 1000 μm diameter were collected by dry-sieving as described elsewhere (Bailey et al., 2012). Nuclease-free gloves were worn while the soil was gently crumbled over a pair of stacked sieves (841 μm and 1000 μm), and 450 field-moist aggregates (13% w/w) were collected using sterile, fine-point tweezers. Mass was determined for each macroaggregate on a AX105DR Delta Range microbalance (Mettler-Toledo, Greifensee, Switzerland), and the macroaggregate was then transferred to a well in a 96-well plate. Aggregate-free wells were randomly distributed on each plate as controls.
Biochemical assays
β-Glucosidase (EC: 3.2.1.21) and ATP were analyzed sequentially on each aggregate (Bailey et al., 2012). Briefly, the β-glucosidase assay (modified from (Saiya-Cork et al., 2002)) was scaled down to 125 μl: 100 μl of acetate buffer (50 mM, pH 5.0) was added to each macroaggregate and the plate agitated. Twenty-five microliters of 4-methylumbelliferyl β-D-glucopyranoside (200 μM) was added to each well, and the plates incubated at 22 °C in the dark for 1 h. The plates were then centrifuged (10 000 × g) for 2 min, the supernatant transferred into a fresh 96-well plate, and 5 μl NaOH (1.0 M) added to stop the reaction. Methylumbelliferone fluorescence was measured immediately, using a Wallac Victor 2 1420 Multilabel Counter (PerkinElmer, Waltham, MA, USA). The pellets from this procedure were kept on ice for the ATP assay.
ATP was assayed with the BacTiter kit (Promega Inc, Madison, WI, USA). Soil pellets were resuspended in 100 μl EDTA (20 mM, pH 7.5) and sonicated for 15 min (Branson Ultrasonics Corp, Danbury, CT, USA). The plate was centrifuged (10 000 × g) for 2 min, and 100 μl of supernatant transferred into a fresh 96-well plate. In a darkened room, 5 μl of MgCl2 (0.4 M) and 100 μl ATP assay reagent were added, the plates shaken gently for 5 min, and luminescence wasmeasured on the Wallac Victor 2 1420 Multilabel Counter. In preparation for DNA extraction, the plates were then centrifuged, the supernatant removed and the pellets frozen at −80 °C.
Grouping of aggregates and DNA extraction
In order to collect enough DNA for 16S pyrosequencing, individual aggregates had to be pooled. The aggregates were sorted by β-glucosidase activity, and aggregates with β-glucosidase activities within 1 s.d. of the mean β-glucosidase activity for the whole set were discarded. The remaining aggregates were randomly sorted into five groups of ‘high activity’ aggregates (HAP) and seven groups of ‘low activity’ aggregates (LAP). The total ATP measured in each aggregate was used as a surrogate for biomass to adjust the groups, such that each group had approximately the same total ATP content. Thus, the final groups were composed of 8–10 aggregates for the low activity class, and 5 groups of 11 aggregates for the high activity class (Table 1). Each group, whether in the high or low activity class, had approximately the same total ATP content (69 fmol).
DNA was extracted from the aggregates using the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA). The selected aggregates of a group were resuspended in equal portions of the PowerSoil buffer for a single reaction. The soil-lysis buffer suspension was then returned to the reaction tube and the kit protocol followed for soil DNA isolation.
Pyrosequencing
The V4 region of the 16S rRNA gene was amplified and sequenced at the Research and Testing Laboratory (Lubbock, TX, USA). Purified DNA from the 12 grouped samples was barcoded and amplified using primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACVSGGGTATCTAAT-3′), and sequenced from the 5′ end of the amplicon (from the barcoded 515F primer) using Roche 454 FLX Titanium reagents (Branford, CT, USA). The sequence data were processed and OTUs identified using mothur v.1.23 (Schloss et al., 2009). Briefly, sequences with ambiguous bases or homopolymers greater than 8 bases were excluded, as were sequences that did not align to the V4 region of the Silva 16S rDNA reference alignment ((Pruesse et al., 2007); http://www.arb-silva.de/), or that were identified as chimeric by UCHIME (http://drive5.com/uchime/). The remaining sequences were aligned to the Silva 16S rDNA reference alignment, assigned to OTUs at ⩾97% identity (with furthest neighbor linkage), and taxonomy assigned using the Ribosomal Database Project reference taxonomy ((Cole et al., 2009); http://rdp.cme.msu.edu/index.jsp). After processing the data for quality, the 12 grouped samples each contained between 33 and 739 sequences with a median length of 222 nucleotides. Randomly subsampling 350 sequences from each grouped sample eliminated two of the low activity samples, and the remaining five low-activity and five high-activity samples were retained for analysis.
Statistical analyses
Alpha and beta diversities: within each activity-level group, we quantified alpha and beta diversities using the ‘numbers equivalents’ framework (Jost, 2007); alpha diversity measures the diversity of a given community and beta diversity measures how variable composition is among communities. Within this framework the parameter q defines the ‘diversity order,’ which determines the degree to which rare or abundant OTUs are emphasized in the calculation of alpha and beta diversities. As q increases from 0 to 2, increasing weight is placed on abundant OTUs. When q=1, equal weights are given to rare and abundant OTUs (Jost, 2007). We use this framework to evaluate how between-activity-level differences in alpha and beta diversity change as abundant OTUs are increasingly emphasized. Following Jost (2007), alpha diversity for a single assemblage is calculated as:

Where  is the relative abundance of the ith OTU in assemblage k, which contains S OTUs. To compare alpha diversity between-activity-level groups, we calculated the mean and s.e.of alpha diversity. Calculation of the mean is described below. For a group with N assemblages, s.e. was found as the s.d. across all N values of qDαk divided by the square root of N.
is the relative abundance of the ith OTU in assemblage k, which contains S OTUs. To compare alpha diversity between-activity-level groups, we calculated the mean and s.e.of alpha diversity. Calculation of the mean is described below. For a group with N assemblages, s.e. was found as the s.d. across all N values of qDαk divided by the square root of N.
To calculate beta diversity for each activity-level group, we used Whittaker’s multiplicative formula, where beta diversity (qDβ) is regional (that is, gamma) diversity (qDγ) divided by mean alpha diversity ( ). Prior to analysis, all assemblages were rarefied to contain 350 sequences, such that all assemblages have equal weight in the calculation of mean alpha diversity. In turn, mean alpha diversity was found as:
). Prior to analysis, all assemblages were rarefied to contain 350 sequences, such that all assemblages have equal weight in the calculation of mean alpha diversity. In turn, mean alpha diversity was found as:

Where N is the number of assemblages (Jost( 2007)). For each activity-level group, gamma diversity was found as:

Where  is the relative abundance of the jth OTU across all assemblages within a group, and Stot is the total number of OTUs observed across all assemblages within a group. Because all communities contained the same number of sequences, this formulation of gamma diversity is identical to that used in Jost (2007).
is the relative abundance of the jth OTU across all assemblages within a group, and Stot is the total number of OTUs observed across all assemblages within a group. Because all communities contained the same number of sequences, this formulation of gamma diversity is identical to that used in Jost (2007).
Greater variation in community composition will be manifest in higher levels of beta diversity (Anderson et al., 2011). The magnitude of beta diversity within an activity-level group is related to the number of OTU combinations that give rise to the observed activity level. Lower beta diversity suggests that community composition is more constrained, such that there are fewer OTU combinations that result in a given level of activity. By comparing beta diversity between the activity-level groups, we can infer whether a change in function is associated with a change in the degree to which community composition is constrained. Further, we compare the difference in beta diversity across diversity orders. Doing so allows us to evaluate whether there is a between-group difference in the degree of constraint and whether this ‘constraint difference’ depends on the degree to which rare versus abundant OTUs are emphasized. For example, if beta diversity in the high-activity group drops below the beta diversity in the low-activity group as diversity order increases, it would provide two inferences: (i) no difference in beta diversity at low diversity orders suggests there is no difference in the number of rare OTU combinations that give rise to high versus low activity levels; and (ii) lower beta diversity in the high activity group at high diversity orders suggests there are fewer combinations of abundant OTUs that result in high activity (and more combinations of abundant OTUs that result in low function).
Compositional dissimilarity within groups: the above described approach provides a single beta diversity value for each activity-level group at each diversity order. This does not allow for significance tests. To compare the level of beta diversity within the low activity group to the beta diversity within the high activity group, we calculated compositional dissimilarity (1-overlap) for all community pairwise comparisons within each activity level group. This was done for the two extremes of the diversity order spectrum (q=0 and q=2). To do so, we used equation 23 in Jost (2007) as implemented in the R function ‘sim.table’ in package ‘vegetarian’. For each diversity order, the dissimilarities were compared across groups using the homogeneity of group dispersions test (Anderson, 2006) (R function ‘betadisper’ in package ‘vegan’, followed byanalysis of variance.).
Community membership and structure comparison: the microbial community membership and structure of the high and low activity samples were compared using analysis of molecular variance, as implemented in mothur. Identification of differentially represented OTUs was done with Metastats (White et al., 2009). Non-metric multidimensional scaling was performed in mothur (‘nmds’ command), using the Bray-Curtis distances between samples, and the resulting ordination visualized in MATLAB (MathWorks Inc., Natick, MA, USA).
Results
β-Glucosidase activity assay and binning
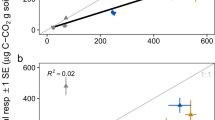
For each aggregate, analysis of β-glucosidase activity was followed by an ATP assay as an estimate of microbial biomass. When the β-glucosidase and ATP measurements were correlated, there was no relationship in single aggregates (Figure 1; R2 was 0.0036). None of the aggregates assayed had β-glucosidase activities below our fluorescence detection; across all aggregates the mean β-glucosidase activity was 74.6 μm methylumbelliferone h−1 per aggregate, and the s.d. was 36.2 μm methylumbelliferone h−1 per aggregate (see Supplementary Figure 1). We used these data to identify the aggregates with native β-glucosidase activities more than 1 s.d. above or below the mean. By this definition, of the 450 aggregates assayed, there were 68 aggregates that classified as ‘low activity,’ and 55 aggregates that classified as ‘high activity.’ To produce enough material for DNA extraction and molecular analysis, aggregates were then pooled, such that each pool had approximately the same combined ATP content, 69 fmol (s.d., 3.9 fmol; Table 1).

β-Glucosidase activities and ATP content in 450 macroaggregates.
The DNA from these high- and low-activity pooled samples was extracted and the bacterial communities profiled by sequencing the V4 region of the 16S rRNA gene. The 12 pooled samples had an average of 432 high-quality sequence reads per sample, but were subsampled to 350 sequences per sample, to avoid bias due to differences in sequencing depth. This subsampling eliminated two low-activity samples with the lowest number of sequences (Table 2). From the 3500 sequences from the remaining 10 samples, 706 OTUs defined at 97% similarity were identified (see Materials and methods), with each sample containing on average 143 OTUs (±15). The ranked, relative abundance of the 706 OTUs, show a typical pattern found in microbial communities, with fewer than 10 OTUs found in >1% relative abundance and a large number of OTUs represented by only one or two sequences (Figure 2).

The rank abundance of the 706 OTUs identified, plotted as a histogram (a) and the traditional Whittaker plot (b).
Alpha and beta diversity of the two activity-level groups
To examine the influence of changing the emphasis from rare to abundant OTUs, we examined alpha and beta diversity over a range of diversity orders. Alpha diversity describes the diversity of the community members within a single sample. Across all diversity orders (see Methods), there was no significant difference in alpha diversity between the two activity-level groups (Figure 3a). Beta diversity was used to describe the similarities and differences in community structures found within pools from the same activity group and also between-activity-level groups. When the diversity order was set to zero, that is, emphasizing rare members, the two groups had effectively indistinguishable levels of beta diversity (Figure 3b), and this was confirmed using the homogeneity of group dispersions test (F1,8=0.05, P=0.8). With increasing diversity order, that is, increasing emphasis on abundant members, beta diversity declined in both groups, and beta diversity of the high-activity group dropped below that of the low-activity group (Figure 3b). This difference was nonsignificant per the homogeneity of group dispersions test (F1,8=1.75, P=0.2).

(a) Alpha diversity as a function of diversity order (given by the parameter q). Higher diversity orders increasingly emphasize abundant OTUs. Dashed lines show mean alpha diversity within each activity-level group. Solid lines encapsulate ±2 s.e., which show overlap between the two groups across all diversity orders; alpha diversity is therefore not significantly different between the two activity-level groups, irrespective of diversity order. (b) Beta diversity as a function of diversity order. The difference in beta diversity between groups is nonsignificant even at diversity order 2.
Community structure of the activity-level groups
We examined the community structure of the two activity-level groups to determine whether there were differentially represented taxonomic groups, which may account for their difference in β-glucosidase activity. A nonparametric analysis of variance on distances between the samples calculated using Bray–Curtis revealed a significant difference between the groups (P<1e-05); the phylogeny-based method UniFrac gave a similar result (unweighted P=0.00710; weighted P<0.00010), as did analyses using additional diversity metrics for community membership and structure (see Supplementary Data Table 2). This separation of the high and low activity groups was significantly correlated with the β-glucosidase activity (P=0.000344), whereas there was no significant correlation based on ATP content, and only marginal correlation with the total mass of the samples (see Supplementary Data Tables 1 and 3, and Figure 2).
The 15 most abundant OTUs account for 1470 of the 3500 (42%) sequences analyzed; Figure 4 shows the relative abundance of these 15 OTUs in the 10 samples, and lists their taxonomic assignments. Two OTUs were identified as significantly differentially abundant between the high and low activity groups: OTU1 was identified as a member of the family Chitinophagaceae and OTU3 was identified as a member of the genus Gemmatimonas. Figure 5 shows the separation of the two activity-level groups based on community structure, visualized in an ordination plot with the differential abundance of the Chitinophagaceae (OTU1; q<1e−06) reflected in the relative size of the data points. Not shown in Figure 5 is OTU3, which was somewhat more abundant in the low activity samples (q=0.014).

Heatmap of the relative abundance of the 15 most abundant OTUs. Significantly differentially represented OTUs are indicated with an arrow.

Non-metric multidimensional scaling (NMDS) ordination of the community structures calculated with Bray–Curtis distances. Symbols representing the high-activity (green) and low-activity (red) samples are scaled to reflect the abundance of the significantly differentially represented OTU1 (q<1e—06), identified as a member of the family Chitinophagaceae. The differential abundance of OTU3 (q=0.014) is not shown; this OTU was somewhat more abundant in the low activity samples.
Discussion
It has been known for some time, that the smaller the sample of soil from which DNA is extracted, the more variable the community profile between samples (Ellingsoe and Johnsen, 2002). This was perceived as a hindrance to a global understanding of community structure for a soil, however, it also suggested that heterogeneities in community composition may occur at fine spatial scales in soils. The research discussed herein uses aggregates less than 1 mm in diameter. One study has suggested that there may be resource gradients operating at this scale that may affect microbial community structures in soils, specifically, roots and residues were measured to have zones of influence less than 2–3 mm (Marschner et al., 2012). Thus, it is conceivable that two irregularly shaped aggregates less than 1 mm in diameter and separated by macropores, could host distinct microbial communities, separated both physically and chemically.
In previous work, we have extracted and sequenced DNA from individual aggregates (Bailey et al., 2013), which depended on the use of whole-genome amplification to provide sufficient DNA for pyrosequencing. In the work presented herein, we chose to isolate a very high number (450) of individual aggregates and used the biomass-normalized distribution of β-glucosidase activity to identify small groups of aggregates with significantly low or significantly high activity, which when pooled would provide sufficient biomass for direct extraction and sequencing of DNA.
The inclusion of ATP in our analyses was an attempt to account for the patchy distribution of soil microorganisms (Pallud et al., 2004), and observed at micron scales in rhizosphere systems (Dandurand et al., 1997). The ATP content would identify aggregates with little or no biomass and that then would be expected to contain minimal β-glucosidase activities; no aggregates in our study fell into such a class. We were also seeking to identify the opposite condition; aggregates with very high ATP contents (and hence high microbial population size) and consequently high β-glucosidase activities because of being very densely populated, which did not occur. Given that the high activity aggregates have ∼fivefold (on average) greater β-glucosidase activities than the low activity aggregates and the aggregates were normalized for active biomass size, our observations suggest that there may be a selection pressure to expand the β-glucosidase competent microorganisms in certain aggregates. This pressure may have been the presence of particular cellulosic substrates derived from organic residues and roots. Residue-C may be incorporated by microbial biomass more than C derived from roots (Marschner et al., 2012), suggesting that colocation of microbes with solid substrate within a niche is important to C dynamics, in contrast to microbial assimilation of root-C, which may occur as soluble or diffusible compounds.
The pyrosequencing yielded a total of just 5185 sequences, which may reflect the low biomass present in the aggregate groups, as well as the challenges associated with extracting high quality DNA from soil. However, we routinely include archived DNA samples from whole soil as well as purified genomic DNA from bacterial isolates as controls, and randomize the samples in the plates sent for sequencing. These control samples (four each of the whole soil and genomic DNA) also yielded relatively low numbers of sequences (data not shown), but analysis of the taxa present was consistent among the samples and with the expected results; thus these controls served to mitigate concerns regarding poor DNA quality from our high- and low-activity aggregate groups as the cause of low sequence yield. Furthermore, upon classifying the pooled sample sequences into 706 total OTUs and assigning each to an appropriate taxon, we observed that the most abundant detected phyla were typical of most soils, including Bacteroidetes, Gemmatomonadetes, Verrucomicrobia, Actinobacteria and Proteobacteria.
Among the OTUs in our high- and low-activity samples, and at the scale interrogated, we found no link between activity level and alpha diversity regardless of diversity order, suggesting that in this system, function is unrelated to the number of rare species and is unrelated to the number of dominant species. We also found no significant relationship between activity level and beta diversity irrespective of diversity order. Although no significant differences were observed, as diversity order increased, high-activity beta diversity did drop below beta diversity of the low-activity group. This trend suggests that, compared with low activity, there may be fewer combinations of abundant OTUs that result in high activity. To clarify whether the trend is biologically meaningful, it would be useful to increase statistical power by using a larger number of samples per activity-level group. The evidence at hand, however, suggests that in our study system there is no relationship between bacterial diversity and ecosystem function, regardless of how bacterial diversity is quantified; expectations are that reducing the scale of a study should enhance our ability to detect linkages between the structure and function (Naeem et al., 2012). We did observe separation of our activity groups, however, the drivers of separation were more subtle than community membership.
Our statistical analyses of the bacterial community present in the high- and low-activity groups revealed that there were differences in the bacterial community structures. Of particular note was the differential abundance of an OTU (OTU1) identified as a member of the family Chitinophagaceae, which was more abundant in the high activity samples. The Chitinophagaceae are members of the phyla Bacteroidetes, which are ubiquitous in soil (Lauber et al., 2009). Currently, two species from the Chitinophagaceae have complete genomic information available: Chitinophaga pinensis and Niastella koreensis, both sequenced as part of the Genomic Encyclopedia of Bacteria and Archaea program (http://img.jgi.doe.gov/cgi-bin/geba/main.cgi; (Wu et al., 2009)). These species are both aerobic organisms isolated from soil samples, and encode several proteins predicted to have β-glucosidase activity (EC: 3.2.1.21). It is possible that cellulytic organisms are latent in aggregates that lack the appropriate substrate or resources for cellulytic activities, but that the presence of substrate or resources allows the members to flourish. This would explain the observation that the differential abundance of OTU1, not its absolute presence or absence, drives the separation of our two groups of aggregates. This hypothesis is also consistent with our observation that β-glucosidase activity had no correlation with total ATP in an aggregate.
There has been evidence that the microbial communities located on the surface of aggregates differ from those in the interior (Ranjard et al., 2000; Mummey and Stahl, 2004), however. the approaches used to determine this relied on surface washing and DNA extraction from aggregate size fractions, rather than an approach using individual aggregates. To examine the soil microbial communities at the pore scale, 13C-labeled substrates were added to soils at various matric potentials, and the incorporation of the label to phospholipid fatty acids was then assessed (Ruamps et al., 2011). This was the first clear evidence that there is a variation in microbial community structure at the pore-scale and that it is not entirely random; again, however, this study analyzed the fatty acid profile of aggregate size fractions, rather than individuals (Ruamps et al., 2011). While significantly advancing our understanding of microbial community structure at the pore scale, these researchers remained unable to deconvolve the contributions of microbial community structure and soil physical structure to C metabolism in soils. Our findings of substantial community overlap in very small groups of aggregates sorted by activities, suggests that a third factor may contribute, and that the presence of resources that permit enhanced activities may be important as well.
It is suggested that fungi are crucial to the development of soil pore structure and the self-organization of microbial communities in soils (Crawford et al., 2012) and in soil C dynamics (Bailey et al., 2002). However, given our constraints on the DNA yield from the aggregate groups, and the knowledge that fungi are not uniquely associated with the enzyme assayed herein, β-glucosidase, we pyrosequenced only 16S rRNA genes, which does not capture fungal sequences. Assay of the fungal community would be informative, particularly in a study of aggregates of broader size ranges, as fungi and bacteria may preferentially dominate different size classes of aggregates, with bacterial biomass dominating in aggregates less than 2 mm, and fungal biomass predominantly associated with aggregates greater than 2 mm (Jiang et al., 2011). It has also been suggested experimentally that bacterial diversity is more significant than fungal diversity in the silt and clay particulate fractions (compared with coarse sands >250 μm) (Poll et al., 2003), such as those that compose the macroaggregates in this study. Nevertheless, accounting for the fungal community and their possible contribution to target soil processes will be an integral part of future studies.
The sheer diversity and abundance of microorganisms suggests that significant functional redundancy occurs in soils (Girvan et al., 2005). Biochemical functionality has been shown to return to a pre disturbance state before community structure does likewise (Berga et al., 2012), and in many studies, community structure never returns to the original state. This begs questions as to the scale at which functional redundancy is active. The observations herein, would suggest that the immediate local condition, that is, the availability of substrate and resources, is exploited by organisms that are present, but that the function assayed, β-glucosidase activity, was predominantly expressed by a specific taxonomic group.
Conclusions
In spite of there being no significant differences in the richness or diversity of the microbial communities associated with high β-glucosidase activities, compared with the communities associated with low β-glucosidase activities, several analyses of variance clearly show that the communities of these two groups differ. The separation of these groups is partially driven by the differential abundances of members of the Chitinophagaceae family. Interestingly, this separation is based on differential abundances, not presence or absence of the OTUs, suggesting that either closely related strains that are not differentiable via 16S sequencing are functionally different, or that the presence of appropriate substrates in the high activity macroaggregates enhances the abundance and activity of competent organisms, which may be present but not stimulated in the low activity macroaggregates.
References
Anderson MJ . (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62: 245–253.
Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL et al (2011). Navigating the multiple meanings of beta diversity: a roadmap for the practicing ecologist. Ecol Lett 14: 19–28.
Bailey VL, Bilskis CL, Fansler SJ, McCue LA, Smith JL, Konopka A . (2012). Measurements of microbial community activities in individual soil macroaggregates. Soil Biol Biochem 48: 192–195.
Bailey VL, Fansler SJ, Bandyopadhyay S, Smith JL, Waters KM, Bolton H . (2010). Direct detection of soil mRNAs using targeted microarrays for genes associated with lignin degradation. Soil Biol Biochem 42: 1793–1799.
Bailey VL, McCue LA, Fansler SJ, Boyanov MI, DeCarlo F, Kemner KM et al (2013). Micrometer-scale physical structure and microbial composition of soil aggregates. Soil Biol Biochem; http://dx.doi.org/10.1016/j.soilbio.2013.02.005.
Bailey VL, Smith JL, Bolton H Jr . (2002). Fungal-to-bacterial ratios in soils investigated for enhanced carbon sequestration. Soil Biol Biochem 34: 997–1007.
Berga M, Szekely AJ, Langenheder S . (2012). Effects of disturbance intensity and frequency on bacterial community composition and function. PLoS One 7: e36959.
Cai P, Huang QY, Zhang XW . (2006). Interactions of DNA with clay minerals and soil colloidal particles and protection against degradation by DNase. Environ Sci Technol 40: 2971–2976.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al (2009). The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Crawford JW, Deacon L, Grinev D, Harris JA, Ritz K, Singh BK et al (2012). Microbial diversity affects self-organization of the soil-microbe system with consequences for function. J R Soc Interface 9: 1302–1310.
Dandurand LM, Schotzko DJ, Knudsen GR . (1997). Spatial patterns of rhizoplane populations of Pseudomonas fluorescens. Appl Environ Microb 63: 3211–3217.
Dokic L, Savic M, Narancic T, Vasiljevic B . (2010). Metagenomic analysis of soil microbial communities. Arch Biol Sci 62: 559–564.
Ellingsoe P, Johnsen K . (2002). Influence of soil sample sizes on the assessment of bacterial community structure. Soil Biol Biochem 34: 1701–1707.
Gans J, Wolinsky M, Dunbar J . (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309: 1387–1390.
Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA . (2005). Bacterial diversity promotes community stability and functional resilience after perturbation. Environ Microbiol 7: 301–313.
Jiang X, Wright AL, Wang X, Liang F . (2011). Tillage-induced changes in fungal and bacterial biomass associated with soil aggregates: a long-term field study in a subtropical rice soil in china. Appl Soil Ecol 48: 168–173.
Jost L . (2007). Partitioning diversity into independent alpha and beta components. Ecology 88: 2427–2439.
Kumaresan D, Stralis-Pavese N, Abell GCJ, Bodrossy L, Murrell JC . (2011). Physical disturbance to ecological niches created by soil structure alters community composition of methanotrophs. Env Microbiol Rep 3: 613–621.
Lauber CL, Hamady M, Knight R, Fierer N . (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microb 75: 5111–5120.
Marschner P, Marhan S, Kandeler E . (2012). Microscale distribution and function of soil microorganisms in the interface between rhizosphere and detritusphere. Soil Biol Biochem 49: 174–183.
McGrath KC, Mondav R, Sintrajaya R, Slattery B, Schmidt S, Schenk PM . (2010). Development of an environmental functional gene microarray for soil microbial communities. Appl Environ Microb 76: 7161–7170.
Morales SE, Holben WE . (2011). Linking bacterial identities and ecosystem processes: can 'omic' analyses be more than the sum of their parts? FEMS Microbiol Ecol 75: 2–16.
Mummey DL, Stahl PD . (2004). Analysis of soil whole- and inner-microaggregate bacterial communities. Microb Ecol 48: 41–50.
Naeem S, Duffy JE, Zavaleta E . (2012). The functions of biological diversity in an age of extinction. Science 336: 1401–1406.
Pallud C, Dechesne A, Gaudet JP, Debouzie D, Grundmann GL . (2004). Modification of spatial distribution of 2,4-dichloro-phenoxyacetic acid degrader microhabitats during growth in soil columns. Appl Environ Microb 70: 2709–2716.
Pietramellara G, Ascher J, Borgogni F, Ceccherini MT, Guerri G, Nannipieri P . (2009). Extracellular DNA in soil and sediment: fate and ecological relevance. Biol Fert Soils 45: 219–235.
Poll C, Thiede A, Wermbter N, Sessitsch A, Kandeler E . (2003). Micro-scale distribution of microorganisms and microbial enzyme activities in a soil with long-term organic amendment. Eur J Soil Sci 54: 715–724.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al (2007). Silva: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Ranjard L, Poly F, Combrisson J, Richaume A, Gourbiere F, Thioulouse J et al (2000). Heterogeneous cell density and genetic structure of bacterial pools associated with various soil microenvironments as determined by enumeration and DNA fingerprinting approach (RISA). Microb Ecol 39: 263–272.
Ruamps LS, Nunan N, Chenu C . (2011). Microbial biogeography at the soil pore scale. Soil Biol Biochem 43: 280–286.
Saiya-Cork KR, Sinsabaugh RL, Zak DR . (2002). The effects of long term nitrogen deposition on extracellular enzyme activity in an Acer saccharum forest soil. Soil Biol Biochem 34: 1309–1315.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541.
van Zyl WH, du Plessis L, Rose SH . (2010). Exploring improved endoglucanase expression in Saccharomyces cerevisiae strains. Appl Microbiol Biotech 86: 1503–1511.
Wang Y, Morimoto S, Ogawa N, Fujii T . (2011). A survey of the cellular responses in Pseudomonas putida KT2440 growing in sterilized soil by microarray analysis. FEMS Microbiol Ecol 78: 220–232.
White JR, Nagarajan N, Pop M . (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 5: e1000352.
Wooley JC, Godzik A, Friedberg I . (2010). A primer on metagenomics. PLoS Comput Biol 6: e1000667.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN et al (2009). A phylogeny-driven genomic encyclopaedia of bacteria and archaea. Nature 462: 1056–1060.
Acknowledgements
This research was funded by the Microbial Communities Initiative LDRD Program at the Pacific Northwest National Laboratory, a multiprogram national laboratory operated by Battelle for the DOE under Contract DE-AC06-76RL01830.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Bailey, V., Fansler, S., Stegen, J. et al. Linking microbial community structure to β-glucosidic function in soil aggregates. ISME J 7, 2044–2053 (2013). https://doi.org/10.1038/ismej.2013.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.87
Keywords
This article is cited by
-
Soil pH indirectly determines Ralstonia solanacearum colonization through its impacts on microbial networks and specific microbial groups
Plant and Soil (2023)
-
Elevated atmospheric CO2 alters the microbial community composition and metabolic potential to mineralize organic phosphorus in the rhizosphere of wheat
Microbiome (2022)
-
Microscale heterogeneity of soil bacterial communities under long-term fertilizations in fluvo-aquic soils
Soil Ecology Letters (2022)
-
Different Degrees of Niche Differentiation for Bacteria, Fungi, and Myxomycetes Within an Elevational Transect in the German Alps
Microbial Ecology (2019)
-
Divergent responses of bacterial activity, structure, and co-occurrence patterns to long-term unbalanced fertilization without nitrogen, phosphorus, or potassium in a cultivated vertisol
Environmental Science and Pollution Research (2019)
