
Abstract
Hydrogenotrophic microbiota have a significant impact on colonic health; however, little is known about their diversity and ecology in situ. Here, molecular-based methods and multivariate analyses were used to examine the abundance and diversity of mucosa-associated hydrogenotrophic microbes in 90 biopsies collected from right colon, left colon and rectum of 25 healthy subjects. Functional genes of all three hydrogenotrophic groups were detected in at least one colonic region of all subjects. Methanogenic archaea (MA) constituted approximately one half of the hydrogenotrophic microbiota in each colonic region. Sulfate-reducing bacteria (SRB) were more abundant than acetogens in right colon, while acetogens were more abundant than SRB in left colon and rectum. MA genotypes exhibited low diversity, whereas SRB genotypes were diverse and generally similar across the three regions within subject but significantly variable among subjects. Multivariate cluster analysis defined subject-specific patterns for the diversity of SRB genotypes; however, neither subject- nor region-specific clusters were observed for the abundance of hydrogenotrophic functional genes. Sequence analyses of functional gene clones revealed that mucosa-associated SRB were phylogenetically related to Desulfovibrio piger, Desulfovibrio desulfuricans and Bilophila wadsworthia; whereas MA were related to Methanobrevibacter spp., Mb. smithii and the order Methanomicrobiales. Together these data demonstrate for the first time that the human colonic mucosa is persistently colonized by all three groups of hydrogenotrophic microbes, which exhibit segmental and interindividual variation in abundance and diversity.
Similar content being viewed by others


Introduction
The human intestinal ecosystem is colonized by a vast number of functionally diverse mutualistic microbes, many of which are abundant and dominant in taxonomic surveys obtained by high-throughput sequencing technologies (Turnbaugh et al., 2009). Less abundant taxa are often undetected by such studies; however, abundance does not necessarily reflect metabolic significance. For example, disposal of H2 gas is fundamental for maintaining efficient microbial fermentation processes, but the microbial groups responsible for this function are present in low abundance (Nakamura et al., 2010). These hydrogenotrophic microbes include acetogens, methanogenic archaea (MA) and sulfate-reducing bacteria (SRB) (Smith and Bryant, 1979; Christl et al., 1992; Strocchi et al., 1994; Bernalier et al., 1996). There is considerable evidence implicating SRB and their end product hydrogen sulfide (H2S) in the pathogenesis of chronic inflammatory disorders of the colon (Roediger et al., 1997; Levine et al., 1998). In addition, H2S is a potent genotoxin (Attene-Ramos et al., 2006), and thus may be linked to sporadic colorectal cancer. Although methane has generally been considered an inert colonic gas, it may influence energy homeostasis of the host through the impact of methanogenesis on the metabolic efficiency of fermentation (Samuel et al., 2007). Furthermore, there is increasing evidence for a link between colonic methane and motility disorders of the intestine, including irritable bowel syndrome (Sahakian et al., 2010).
The colonic microbiota has most often been characterized from stool samples, which do not accurately reflect mucosa-associated microbiota (Zoetendal et al., 2002; Eckburg et al., 2005). Consequently, relatively little is known about the diversity and ecology of mucosa-associated hydrogenotrophic microbes for any mammalian species. Two molecular-based studies demonstrate that proximal colonic (Zinkevich and Beech, 2000) and rectal (Fite et al., 2004) mucosae are persistently colonized by SRB. Relatively limited MA diversity has been detected, with only Methanobrevibacter smithii and Methanosphaera stadtmanae cultured from stool (Miller et al., 1982; Miller and Wolin, 1985) and also predominant in molecular surveys (Eckburg et al., 2005; Zhang et al., 2009). More recently, Oxley et al. (2010) reported the presence of Mb. smithii, Mb. arboriphilus and Msp. stadtmanae 16S rRNA gene sequences in sigmoid colon mucosa.
The present study characterized the abundance and diversity of mucosa-associated hydrogenotrophic microbes in biopsies from three colonic regions taken from healthy subjects. Ribosomal 16S rRNA or functional genes of acetogens, MA and SRB were analyzed by quantitative PCR (qPCR) and molecular fingerprinting. Phylogenetic affiliations of mucosal SRB and MA were further investigated by clone library analyses. Replicate biopsies were also studied to evaluate the extent of microanatomical variation in hydrogenotrophic microbiota.
Materials and methods
Sample collection and preparation
Colonic biopsies were obtained from 20 healthy subjects (12 women and 8 men) with an age range of 47–64 years undergoing routine colonoscopy at Carle Foundation Hospital (Urbana, IL, USA). None of the recruited subjects had taken antibiotics for at least 8 weeks before sample collection. The biopsies were collected during colonoscopy following standard bowel cleansing methods, snap frozen in liquid nitrogen and stored at −80 °C until DNA extraction was performed. Sixty biopsies were collected from right and left colon and rectum. A duplicate biopsy from each location was collected for confirmation of normal tissue by the Carle Pathology Services Laboratory (Urbana, IL, USA). Also, replicate colonic biopsies <1 cm apart were obtained from right colon, left colon and rectum of five additional healthy subjects. Procedures related to collection and use of tissue from human subjects, including informed consent of participants, were reviewed and approved by the Institutional Review Boards of the University of Illinois at Urbana-Champaign and Carle Foundation Hospital. Demographic information including age, gender, race and endoscopic findings is summarized in Supplementary Table S1.
DNA extraction and PCR amplification
Genomic DNA was extracted from biopsies using a commercial kit (QIAamp DNA Stool Mini Kit; Qiagen, Valencia, CA, USA) following Zoetendal et al. (2006). Mechanical cell disruption was not used to avoid excess extraction of eukaryotic DNA, which can interfere with PCR amplification of 16S rRNA genes (Huys et al., 2008; Bakke et al., 2011). The absence of mechanical cell disruption did not affect either the quantity or the quality of microbial DNA isolated as verified by direct comparison of the two approaches with a subset of biopsies (Carbonero et al., in preparation). PCR amplification was performed with primers targeting genes specified in Supplementary Table S2 in 25 μl reactions containing 2.5 μl of 10 × buffer, 2.5 μl of BSA (0.1 mg ml−1), 2 μl of 2.5 mM dNTP, 1 μl of each primer (25 μM), 2 μl (0.75 units) of hot start Taq polymerase (Takara, Shiga, Japan), 1 μl of DNA template, and brought to volume with PCR-grade water. Amplifications were performed by initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturation at 94 °C for 1 min, primer annealing at the specific temperature (Table 2) for 1 min and extension at 72 °C for 1 min, with a final extension at 72 °C for 7 min.
All PCR steps included negative and positive controls (DNA extracted from pure cultures). Amplicons were checked on agarose gel electrophoresis using ethidium bromide fluorescent dye. Triplicate PCR amplifications were performed for each sample, pooled PCR products were gel purified using the QIAquick gel extraction kit (Qiagen).
qPCR analysis of hydrogenotrophic functional genes and SRB genera
Real-time qPCR was performed on all colonic DNA extracts (20 subjects+replicated biopsies from the five additional subjects; A–E, Supplementary Table S1) using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Both primers ACSF1/ACSR1 (Gagen et al., 2010) and FTHFSf/FTHFSr (Leaphart and Lovell, 2001) targeting functional genes acs and fhs of acetogens were used (Supplementary Table S2). Primers ME1/ME2 (Hales et al., 1996) and DSR1fdeg/DSR4rdeg (Leloup et al., 2007) were used for mcrA and dsrA genes, respectively (Supplementary Table S2). Primer pairs targeting 16S rRNA genes of Desulfobacter, Desulfobulbus, Desulfotomaculum/Desulfosporosinus and Desulfovibrio were used to quantify SRB genera (Daly et al., 2000; Supplementary Table S2). Samples were run on a 7900HT Fast Real-Time PCR System (Applied Biosystems) using a dissociation curve. Standard curves were determined simultaneously using plasmids containing dsrA, mcrA and fhs or diluted PCR products from reference strains for acs and the 16S rRNA genes.
Nested PCR amplification of Desulfovibrio and Archaea 16S rRNA genes
Biopsy DNA was used as a template for PCR amplification using universal primers GM3f/GM4r targeting the 16S rRNA gene of the domain Bacteria (Muyzer et al., 1995), which was then subjected to a second round of amplification (610 bp) using 16S rRNA gene primers DSV230f/DSV838r specific for Desulfovibrio spp. (Daly et al., 2000).
For nested PCR of Archaea, the first amplification was carried out using universal primers that target the 16S rRNA gene of the Archaea domain (Hallam et al., 2003), and products were then subjected to a second round of amplification using an internal set of specific Archaea 16S rRNA gene primers (Grosskopf et al., 1998).
PCR amplification of dissimilatory sulfite reductase and methyl-coenzyme M reductase genes
For nested PCR of dsrA, DNA extracted from biopsy tissues samples was used as a template for direct PCR amplification using primers DSR1F (Wagner et al., 1998) and DSRAB-R-b (Schmalenberger et al., 2007). Subsequently, these PCR amplicons were subjected to a second round of amplification using internal-specific primers 1FI (Dhillon et al., 2003) and DSR10R (Barneah et al., 2007).
For nested PCR of mcrA, direct PCR amplification was carried out using primers ME1 (Hales et al., 1996) and MCRr (Springer et al., 1995). Direct PCR products were then subjected to a second round of amplification using an internal set of specific mcrA gene primers ME2 (Hales et al., 1996) and MCRrb (Springer et al., 1995).
Terminal restriction fragment length polymorphism analysis
Genomic diversity of 16S rRNA (Desulfovibrio, Archaea) and functional genes (dsrA and mcrA) was examined in the initial set of 20 subjects (1–20, Table 1) by terminal restriction fragment length polymorphism (T-RFLP; Liu et al., 1997). Forward primers for nested amplification of each target gene were 5′-end labeled with 6-carboxyfluorescein (6-FAM; Integrated DNA Technologies, Coralville, IA, USA). For each microbial gene target, T-RFLP analyses were performed using two independent restriction enzymes. T-RFLP analysis of Desulfovibrio spp. 16S rRNA genes was carried out using 10 μl aliquots (3.75 ng of DNA per μl) of the amplicons produced by nested PCR. These DNA products were cleaved for 6 h in a water bath at 37 °C with 2.5 units of restriction endonuclease following the manufacturer recommendations (NE Biolabs, Ipswich, MA, USA). HpyCH4IV and ScrfI endonucleases were used for digestion of Desulfovibrio spp. 16S rRNA gene amplicons. T-RFLP analyses of Archaea 16S rRNA, dsrA and mcrA were performed as described above with the following exceptions: For the analysis of Archaea 16S rRNA genes, 10 μl aliquots (7.5 ng of DNA per μl) of nested PCR amplicons were digested with BfaI and Hpy188I endonucleases (NE Biolabs). For the analysis of dsrA, 10 μl aliquots (6.0 ng of DNA per μl) of nested PCR amplicons were digested with Sau96I and BstUI endonucleases (NE Biolabs). For mcrA, 10 μl aliquots (6.0 ng of DNA per μl) of nested PCR amplicons were digested with DdeI and Hpy188III endonucleases (NE Biolabs).
DNA fragment analysis was performed on the ABI Prism 3730xl Analyzer using GeneScan Liz600 marker (Applied Biosystems) as a size standard. Terminal restriction fragment (TRF) profiles in each sample were produced using GeneMapper software (version 3.7; Applied Biosystems). For TRF profiles of 16S rRNA genes, terminal fragments smaller than 50 bp and lower than 100 arbitrary fluorescence units were considered background noise and manually removed from the TRF profile in each sample. For TRF profiles of dsrA and mcrA, terminal fragments smaller than 10 bp were manually removed from the TRF profile in each sample. Background noise found in the T-RFLP analysis of 16S rRNA gene amplicons produced from DNA extracted from pure cultures (positive controls) was also removed from the TRF profiles in each sample.
Statistical analyses of microbial profiles
TRF profiles were aligned on the basis of TRF lengths and individual peak areas using the moving average algorithm included in the T-Align software (Smith et al., 2005), resulting in the generation of data sets of aligned TRFs. Profiles obtained with two independent restriction analyses (endonucleases) were concatenated to form a collective data set, and the resulting output files were used in multivariate statistical analyses using an inferential statistic analysis (non-parametric multivariate ANOVA) for the quantitative assessment of microbial profiles and the use of a non-metric technique, non-metric multidimensional scaling (NMDS) analysis. The results of the non-metric multidimensional scaling ordinations were corroborated by a linear model, principal coordinate analysis, and by a non-parametric clustering technique, multivariate cluster analysis (MCA). For analyses of presence–absence of TRFs, the Kulczynski index was used, whereas analyses of relative abundance-based data were carried out using the Morisita index (Anderson et al., 2011). Interindividual variability within the three colonic regions was estimated using pairwise similarities between right colon, left colon and rectum using those indices. All multivariate statistical analyses were performed with the PAST software package (Hammer et al., 2001). In addition, ANOVA and Fisher′s Protected Least Significant Difference test were used to compare intergroup variability among right colon, left colon and rectum. These analyses were carried out using SAS software (Statview, Version 5.0.1; SAS Institute, Cary, NC, USA).
Phylogenetic analysis of dissimilatory sulfite reductase and methyl-coenzyme M reductase genes
Biopsy samples with the highest diversity of dsrA or mcrA genes (as determined by T-RFLP) were selected, and genomic DNA from these samples was used as a template for nested PCR (without primers 5′-end labeled with 6-FAM). Purified PCR products from the colonic biopsies were pooled and cloned using the TOPO-TA kit (Invitrogen, Carlsbad, CA, USA) as per the manufacturer recommendations. Transformants were propagated in LB medium overnight and used for plasmid DNA extraction using the QIAprep Spin MiniPrep Kit (Qiagen). Sequencing using the M13F (−21) and M13R (−48) primers was performed with an ABI 3730XL capillary sequencer (Applied Biosystems). Chromatogram checks, trimming and assembling of sequences were performed with Sequencher 4.9 (Gene Codes Corporation, Ann Arbor, MI, USA).
Consensus nucleotide sequences were used for BLAST analysis (Altschul et al., 1997) against the non-redundant nucleotide database from GenBank. The 20 best hits (E-value <3E-120) for each clone sequence were retrieved and used for phylogenetic analyses. Nucleotide sequences were aligned using ClustalW. Poorly aligned regions (5′- and 3′-flanking segments) were manually removed, and high quality alignments were used for phylogenetic tree reconstructions using the maximum parsimony method. The statistical significance of branch order was estimated by the generation of 500 replications of bootstrap re-sampling of the originally aligned nucleotide sequences. These phylogenetic analyses were conducted with MEGA4 software (Tamura et al., 2007).
Results
Quantification of acs, fhs, mcrA and dsrAB in right colon, left colon and rectal mucosa
Estimates of the abundance of acs, fhs, mcrA and dsrAB gene copies per gram of colonic tissue are shown in Figure 1. Functional genes of all three hydrogenotrophic groups were detected in all colonic regions from all subjects with the exception of mcrA. The acetogenic genes acs and fhs were detected in all biopsies with gene copy numbers ranging from 1.8 × 103 to 8.8 × 106 and from 9.8 × 103 to 3.8 × 107 per gram tissue, respectively. All subjects also harbored significant mcrA gene copy numbers in at least one colonic region ranging from 3.0 × 102 to 4.5 × 109. DsrAB gene copy numbers ranging from 1.8 × 102 to 1.4 × 109 per gram were detected for all biopsies from all subjects. The dsrAB and mcrA genes exhibited the greatest variation in abundance among subjects, whereas the abundance of acs and fhs gene copies was generally in the same order of magnitude (104–105) across colonic regions for all subjects. Overall, the functional gene data indicate that MA constituted approximately one half of the hydrogenotrophic microbiota in each colonic region (Table 1). SRB were more abundant than acetogens in right colon, while acetogens were more abundant than SRB in left colon and rectum. Multivariate statistical analysis of the combined abundance of acs, fhs, mcrA and dsrAB copy numbers via the Morisita index did not reveal coherent clustering among the 60 biopsies by colonic region or subject (Supplementary Figure S1).

Quantification of acs, fhs, mcrA and dsrAB in right colon, left colon and rectal mucosa. Estimates of the abundance of acs, fhs, mcrA and dsrAB gene copies per gram of colonic tissue were determined by real-time PCR with a PCR product or plasmid dilution standard curve of the corresponding functional gene. Error bars indicate the s.e.m. of three technical replicates. MCA of the abundance of the four functional genes is presented in Supplementary Figure S1.
Quantification of SRB genera in right colon, left colon and rectal mucosa
Estimates of the abundance of Desulfobacter, Desulfobulbus, Desulfotomaculum/Desulfosporosinus and Desulfovibrio 16S rRNA gene copies per gram of colonic tissue are shown in Figure 2. Desulfobacter and Desulfobulbus were detected in all 60, Desulfovibrio in 56/60 and Desulfotomaculum in 42/60 biopsies. Desulfotomaculum was the only SRB genus not detected in all subjects, being absent in three (S1, S4 and S12). When present, the abundance of Desulfovibrio 16S gene copy numbers was always greatest (104–107) followed by Desulfobacter and Desulfobulbus (103–106) and finally Desulfotomaculum (102–104). The rectum harbored the least diversity in SRB genera (Figure 2). The total signal for the four genera was roughly 10 times lower than the total dsrAB signal (Table 2). On average, Desulfovibrio was the most abundant in the right colon and Desulfobulbus in the rectum (Table 2). Regional differences were not observed for the abundance of Desulfobacter and Desulfotomaculum.

Quantification of SRB genera in right colon, left colon and rectal mucosa. Estimates of the abundance of Desulfobacter, Desulfobulbus, Desulfotomaculum/Desulfosporosinus and Desulfovibrio 16S rRNA gene copies per gram of colonic tissue were determined by real-time PCR with a PCR product dilution series standard curve of genomic DNA from corresponding type strains. Error bars indicate the s.e.m. of three technical replicates.
Genotypic diversity of mucosa-associated Desulfovibrio spp.
The nested PCR approach revealed that all 20 of the initial set of subjects harbored mucosa-associated Desulfovibrio spp. populations in at least one colonic region. The percentages of biopsy tissues positive for Desulfovibrio spp. in right colon, left colon and rectum were 95, 90 and 100, respectively. Thirteen Desulfovibrio spp. TRFs were obtained with HpyCH4IV and thirty-four with ScrfI. The mean abundance of individual Desulfovibrio spp. TRFs with ScrfI across the three colonic regions is shown in Figure 3a. Three major TRFs constituted >75% of the total profiles and the three colonic regions generally possessed similar TRF composition. Broadly the Desulfovibrio spp. TRFs formed four major groups represented in a minimum of three subjects for each (Figure 3a).

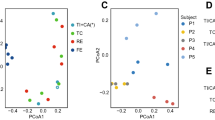
Individual T-RFLP profiles of Desulfovibrio 16S rRNA and dsrA gene sequences. A nested PCR T-RFLP approach was used to examine diversity in Desulfovibrio spp. 16S rRNA genes in mucosal biopsies from right colon (RC), left colon (LC) and rectum (Re). Relative abundance is demonstrated by terminal fragments recovered from the T-RFLP analysis using ScrfI endonuclease for Desulfovibrio 16S rRNA (a). Only TRFs that were present in at least two regions and representing >1% of the total profile signal are presented. Subjects are grouped according to similarities of TRF profiles. MCA of Desulfovibrio spp. 16S rRNA gene profiles across RC (red), LC (green) and Re (blue) was performed from the TRF profiles using Kulczynski (b) and Morisita (c) similarity indices. Cophenetic correlation coefficient (Coph Corr) values are shown. A similar nested PCR T-RFLP approach was used to examine diversity in dsrA using Sau96I endonuclease (d). MCAs of dsrA T-RFLP profiles are presented in Supplementary Figure S2.
Statistical comparisons by means of ANOVA and Fisher′s Protected Least Significant Difference test indicated that right colon harbored significantly more homogenous Desulfovibrio spp. TRFs than left colon (P=0.0059) based on the Kulczynski index; however, the three colonic regions were shown to harbor comparable variability in Desulfovibrio spp. TRFs using the Morisita index (P>0.05). Furthermore, pairwise similarities between these colonic sections using both indices revealed that right colon, left colon and rectum possess comparable variability in Desulfovibrio spp. TRFs (P>0.05). Similar results were obtained using multivariate ANOVA with both indices. MCA confirmed the absence of regional-specific Desulfovibrio spp. TRF patterns. However, subject-specific clusters were observed for Desulfovibrio spp. TRF patterns (Supplementary Figures S2A and B). These ordination patterns were confirmed by non-metric multidimensional scaling and principal coordinate analysis.
The diversity of mucosa-associated Desulfovibrio 16S rRNA genes was also analyzed in replicate colonic biopsies. Specifically, mucosa-associated Desulfovibrio spp. communities clustered (>60% similarity) mainly by subject using presence–absence data (Kulczynski index); only subject E exhibited contrasting TRF profiles (Supplementary Figure S3A). Statistical analysis of Desulfovibrio spp. profiles using TRF abundance data (Morisita index) revealed that mucosa-associated Desulfovibrio spp. communities clustered (>70% similarity) by subject and colonic site (Supplementary Figure S3B). Desulfovibrio spp. profiles from subjects A and B formed a distinct cluster with two exceptions, and right colon and left colon of subject C formed a distinct cluster. Rectal Desulfovibrio spp. communities from four subjects clustered together rather than by subject. The replicate samples of only five of fifteen biopsy pairs clustered together, and these matches were not coherent for the two statistical indices (Supplementary Figure S3). This observation is consistent with microheterogeneity in Desulfovibrio spp. communities within colonic regions.
Diversity of dsrA genes of mucosa-associated microbes
The nested PCR approach revealed that 19 out of 20 subjects harbored mucosa-associated dsrA sequences in at least one region of the colon. The percentages of biopsy tissues positive for dsrA gene amplification in right colon, left colon and rectum were 90, 80 and 80, respectively. The dsrA TRF profiles were less diverse than the Desulfovibrio spp. 16S TRFs. Fourteen SRB TRFs were recovered with BstUI and sixteen TRFs with Sau96I. Mean abundances of dsrA TRFs across the three colonic tissues from Sau96I are shown in Figure 3d. Broadly, the dsrA TRFs formed one major (14/20 subjects) and two minor groups, which were consistent across colonic regions (Figure 3d).
Statistical comparisons by means of ANOVA and Fisher′s Protected Least Significant Difference test using the Kulczynski index revealed that right colon is colonized significantly by more homogenous SRB populations than rectum (P=0.0452); however, the three colonic regions harbored comparable variability using the Morisita index (P>0.05). Furthermore, pairwise similarities between these colonic sites using either index revealed that right colon, left colon and rectum possess comparable variability in SRB populations (P>0.05). Similar results were obtained using multivariate ANOVA with both indices. MCA confirmed the absence of regional-specific dsrA spp. TRF patterns. However, subject-specific clusters were observed for dsrA spp. TRF patterns (Supplementary Figures S2A and B).
Diversity of mucosa-associated Archaea populations
Forty percent of the twenty subjects harbored mucosa-associated Archaea populations in at least one region of the colon. Twenty percent of the subjects were positive for Archaea 16S rRNA gene amplification for all of the three regions. Eight Archaea TRFs were recovered with BfaI and ten with Hpy188I. The mean abundances of Archaea TRFs across colonic sites for Hpy188I are shown in Figure 4a. There were 10 (Hpy188I) or 8 (BfaI) major TRFs and the TRF composition overlapped except for subject 7 left colon. Distinct TRF profiles were observed for the two individuals (2 and 6) that were positive for Archaea for the three colonic sites.

Individual T-RFLP profiles of Archaea 16S rRNA and mcrA gene sequences. A nested PCR T-RFLP approach was used to examine diversity in Archaea 16S rRNA and mcrA genes in mucosal biopsies from right colon (RC), left colon (LC) and rectum (Re). Relative abundance is demonstrated by terminal fragments recovered from the T-RFLP analysis using Hpy188III endonuclease for Archaea 16S rRNA (a) and mcrA (d). Each color represents a terminal fragment and its proportion within each chart represents relative abundance. Regional similarities were estimated by means of MCA across RC (red), LC (green) and Re (blue) using Kulczynski (b) and Morisita (c) similarity indices.
Ordinations derived from the Kulczynski (Figure 4b) and Morisita (Figure 4c) indices revealed two major clusters with the rectal TRF profile of subject 5 being an outlier. The different colonic regions of subjects 2 and 6 formed coherent and distinct clusters corresponding to these two major clusters.
Diversity of methyl-coenzyme M reductase gene of mucosa-associated microbes
In contrast to Archaea 16S rRNA, only 20% of the subjects were positive for mcrA amplification in at least one region of the colon. Two mcrA TRFs were recovered with DdeI and four with Hpy188III, and only one biopsy (RC) from subject 6 exhibited that diversity. The mean abundances of mcrA TRFs across colonic sites for Hpy188III are shown in Figure 4d.
Phylogenetic analysis of dissimilatory sulfite reductase and methyl-coenzyme M reductase genes
Twenty-one dsrA gene clones were obtained from the sixty initial colonic biopsies. On average, the recovered mucosa-associated dsrA clones were 98.5% homologous to dsrA genes archived in GenBank. Phylogenetic tree reconstruction (Figure 5a) using maximum parsimony and statistical significance of branch order (500 replications of bootstrapping) revealed that mucosa-associated SRB were genetically related to Desulfovibrio piger (three clones), Desulfovibrio. desulfuricans (three clones), uncultured SRB (six clones) and Bilophila wadsworthia (nine clones).


Phylogenetic affiliations of dsrA and mcrA clone sequences. Twenty-one clones of dsrA genes and nineteen clones of mcrA genes were recovered from colonic biopsies and sequenced. Phylogenetic tree reconstructions for dsrA (a) and mcrA (b) using the maximum parsimony and statistical significance of branch order (500 replications of bootstrapping) are shown. Red symbols represent the clones obtained in the present study. The scale bar represents the number of nucleotide substitutions per site.
Nineteen mcrA gene clones were recovered from these colonic biopsies. On average, the mucosa-associated mcrA clones were 97.2% homologous to mcrA genes archived in GenBank. Maximum parsimony phylogenetic tree reconstruction (Figure 5b) and statistical significance of branch order (500 replications of bootstrapping) revealed that the mucosa-associated MA were genetically related to uncultured Methanobrevibacter sp. (15 clones), Methanoculleus chikugoensis (3 clones) and Mb. smithii (1 clone).
The dsrA sequences are deposited in GenBank under accession numbers GU180102 through GU180122 and mcrA sequences under accession numbers GU180123 through GU180141.
Abundance of mucosa-associated hydrogenotrophic microbiota in replicate colonic biopsies
Estimates of the abundance of acs, fhs, mcrA and dsrAB gene copies per gram of colonic tissue for the thirty replicate biopsies from three colonic sites of five additional subjects are shown in Figure 6a. Consistent with observations from the set of 60 biopsies from 20 subjects, functional gene sequences for each of the hydrogenotrophic groups were present in at least one colonic region for the five additional subjects. Again, MA abundance was the most variable in the set of replicate biopsies ranging from not detected to 108 mcrA gene copy numbers per gram. Statistical analysis of the combined abundance of acs, fhs, mcrA and dsrAB copy numbers via the Morisita index did not reveal coherent clustering among the replicate biopsies by colonic region or subject (Figure 6b).

Quantification of acs, fhs, mcrA and dsrAB in replicate biopsies from five additional subjects. Estimates of the abundance of acs, fhs, mcrA and dsrAB gene copies per gram of colonic tissue were determined by real-time PCR with a corresponding PCR product or plasmid dilution standard curve (a). Error bars indicate the s.e.m. of three technical replicates. MCA of acs, fhs, mcrA and dsrAB across the RC (red), LC (green) and Re (blue) was performed using the Morisita similarity index (b). Cophenetic correlation coefficient (Coph Corr) values are shown.
Discussion
This study provides the first molecular characterization of low-abundant hydrogenotrophic microbes that are associated with the colonic mucosa of healthy humans. These data confirm that SRB populations are ubiquitous and found associated with the mucosa throughout the colon. Moreover, these data are the first to demonstrate that mucosa-associated MA and acetogens are also prevalent, with all 25 subjects examined generating a signal for related functional genes in at least one colonic region. In addition, this study confirms that Desulfovibrio is generally the most abundant mucosa-associated SRB genus. Consistent with other microbial groups in the colonic ecosystem, both SRB and MA genotypes were highly variable among subjects and without clear region-specific patterns. Statistical analyses supported clustering of particular SRB genotypes by subject but not by colonic region.
Cultivation-based studies have estimated that the number of acetogens ranges from 102 to 108 CFU per gram human feces (Dore et al., 1995; Bernalier et al., 1996). Acetogenesis is thought to be a relatively less important hydrogenotrophic pathway in the colon because the oxidation of H2 by methanogenesis or sulfate reduction is thermodynamically more favorable than reductive acetogenesis (Nakamura et al., 2010). However, recent metagenomic data indicated that genes involved in reductive acetogenesis were more abundant than methanogenesis and sulfate respiration genes (Rey et al., 2010). Our data indicate that mucosal acetogens are present in numbers comparable to MA and SRB. Thus, these data stress the need to further define the metabolic contributions of reductive acetogens to hydrogen disposal in the colonic mucosa.
Both the qPCR and nested PCR data indicate persistent colonization of SRB throughout the colon and Desulfovibrio spp. in particular. These data are consistent with previous reports using cultivation (Macfarlane et al., 1992) as well as Desulfovibrio spp. 16S rRNA (Fite et al., 2004) and functional gene (Zinkevich and Beech, 2000) primers. Unexpectedly, dsrA genes were less diverse than Desulfovibrio spp. 16S rRNA genes, which might indicate that the dsrA primers failed to amplify all of the genetic diversity. In this regard, subsequent efforts to obtain a complete census of mucosal SRB would benefit from the use of more degenerate dsrA primers.
Heretofore, colonic SRB had been characterized mainly by culture-based studies using stool or colonic contents as inocula (Nakamura et al., 2010). For example, Gibson et al. (1993) cultivated SRB from stool of healthy subjects that were classified as Desulfovibrio spp., Desulfobacter spp., Desulfobulbus spp. and Desulfotomaculum spp. Desulfovibrio spp. are typically considered to be the predominant SRB genus in the human colonic ecosystem based on a limited number of culture-based studies (Loubinoux et al., 2002). The present data fully corroborate previous reports on the predominance of Desulfovibrio spp. as well as the presence of other SRB genera including Desulfobulbus spp. and Desulfobacter spp., which were consistently detected throughout the colon; Desulfotomaculum/Desulfosporosinus exhibited a more irregular distribution.
Based on the phylogenetic analysis of dsrA clones, mucosa-associated SRB populations included D. piger, D. desulfuricans, uncultured Desulfovibrio spp. and B. wadsworthia. B. wadsworthia is a member of the mutualistic microbiota but has also been linked to gangrenous and perforated appendicitis in humans (Urban et al., 2004). This genus does not reduce sulfate but can use sulfite as a terminal electron acceptor during taurine metabolism in anaerobic respiration (Laue et al., 2001). It is important to acknowledge that the presence of SRB does not necessarily reflect the occurrence of sulfate reduction. Moreover, it is not possible to reach conclusions on the activity of mucosal SRB from the present molecular data. Nonetheless, evidence for persistent colonization of the colonic mucosa with SRB stresses the importance of gaining knowledge on their in situ metabolic activities.
To our knowledge, the present data provide the first direct evidence for widespread colonization of the colonic mucosa with MA. A mcrA signal was detected in at least one colonic region for all of the 25 subjects examined. In addition, MA were the predominant hydrogenotrophic group throughout the colon. Based on a breath methane concentration of 1 p.p.m. or greater, the percentage of methane producers varies significantly in humans of different ethnic groups, ranging from 34% to 87% (summarized in Levitt et al., 2006). Although breath methane reliably reflects the abundance of colonic methanogens, non-methanogenic subjects, as thereby defined, often harbor a relatively lower number of stool MA (El Oufir et al., 1996). One study, based on a fortuitous measurement of colonic gases, suggested that methane was predominantly produced in the left colon (Bond et al., 1971). Culture-based data consistent with the abundance of MA increasing in a proximal to distal gradient have also been reported (Macfarlane et al., 1992). To a certain extent, our observations concur with this gradient, as mcrA abundance increased along the colon to comprise two thirds of the hydrogenotrophic gene content in the rectum. Archaeal diversity (16S rRNA gene TRFs) was greater than MA diversity (mcrA TRFs). This observation indicates significant colonization by non-MA, which is in accordance with previous detection of non-MA from colonic mucosa (Oxley et al., 2010). Alternatively, it is possible that MA harboring the mrtA functional gene, such as Msp. stadtmanae, were not detected by the nested PCR approach.
Culture- and molecular-based studies to date indicate that Mb. smithii is the predominant methanogen in the human colon (Miller et al., 1982; Weaver et al., 1986; Eckburg et al., 2005; Scanlan et al., 2008; Zhang et al., 2009). Msp. stadtmanae, also a member of the order Methanobacteriales, was isolated from human stool at a lower abundance (Miller and Wolin, 1985). Phylogenetic analysis of the mcrA clones recovered from colonic biopsies revealed that mucosa-associated MA were related to Mb. smithii, uncultured Methanobrevibacter spp. and one clade belonging to the Methanomicrobiales order and distantly related to the Methanoculleus genus. The presence of these sequences confirms that MA lineages other than the two most commonly described are also present in the human colon. Specifically, mcrA and 16S rRNA gene sequences related to Methanosarcinales (Scanlan et al., 2008) and to a putative new order related to Thermoplasmatales (Mihajlovski et al., 2008) have been detected in stool.
The abundance of SRB and MA was highly variable among subjects and without clear host or region-specific patterns. On the other hand, both Desulfovibrio spp. and dsrA TRF profiles generally clustered by subject. Replicate biopsies collected <1 cm apart did not cluster by host or colonic site for the abundance of hydrogenotrophic functional genes. Thus, the composition of hydrogenotrophic microbiota may be individualized, similar to mutualistic microbiota in general; while the abundance of the three groups appears to vary both regionally and in a microanatomical scale. It should be noted that the bowel cleansing preparation may have modified loosely adherent microbiota, which might have led to underestimation of the diversity or abundance of the microbial groups examined. It is not feasible to sample the right or transverse colon of healthy subjects without bowel cleansing; thus, it would be difficult to empirically address these potential biases. Regardless, standardized methodologies were used for the collection and analysis of all biopsy samples validating the relative differences observed.
In summary, the present data from three colonic regions of 25 human subjects provide clear evidence for coexistence of acetogens, SRB and MA, indicating that these three groups are not mutually exclusive in healthy mucosa. These baseline data provide a framework to determine the extent to which dysbiosis in hydrogenotrophic microbiota may be linked to colonic disorders. Moreover, the evidence for microheterogeneity highlights the need to define spatial scales of microbial hydrogenotrophy relevant to disease etiology in the human colon.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL et al. (2011). Navigating the multiple meanings of beta diversity: a roadmap for the practicing ecologist. Ecol Lett 14: 19–28.
Attene-Ramos MS, Wagner ED, Plewa MJ, Gaskins HR . (2006). Evidence that hydrogen sulfide is a genotoxic agent. Mol Cancer Res 4: 9–14.
Bakke I, De Schryver P, Boon N, Vadstein O . (2011). PCR-based community structure studies of bacteria associated with eukaryotic organisms: a simple PCR strategy to avoid co-amplification of eukaryotic DNA. J Microbiol Methods 84: 349–351.
Barneah O, Ben-Dov E, Kramarsky-Winter E, Kushmaro A . (2007). Characterization of black band disease in Red Sea stony corals. Environ Microbiol 9: 1995–2006.
Bernalier A, Lelait M, Rochet V, Grivet JP, Gibson GR, Durand M . (1996). Acetogenesis from H2 and CO2 by methane- and non methane-producing human colonic bacterial communities. FEMS Microbiol Ecol 19: 193–202.
Bond Jr JH, Engel RR, Levitt MD . (1971). Factors influencing pulmonary methane excretion in man. An indirect method of studying the in situ metabolism of the methane-producing colonic bacteria. J Exp Med 133: 572–588.
Christl SU, Gibson GR, Cummings JH . (1992). Role of dietary sulphate in the regulation of methanogenesis in the human large intestine. Gut 33: 1234–1238.
Daly K, Sharp RJ, McCarthy AJ . (2000). Development of oligonucleotide probes and PCR primers for detecting phylogenetic subgroups of sulfate-reducing bacteria. Microbiology 146: 1693–1705.
Dhillon A, Teske A, Dillon J, Stahl DA, Sogin ML . (2003). Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl Environ Microbiol 69: 2765–2772.
Dore J, Pochart, P Bernalier A, Goderel I, Morvan B, Rambaud JC . (1995). Enumeration of H2-utilizing methanogenic Archaea, acetogenic and sulfate-reducing bacteria from human feces. FEMS Microbiol Ecol 17: 279–284.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al. (2005). Diversity of the human intestinal microbial flora. Science 308: 1635–1638.
El Oufir L, Flourie B, Bruley des Varannes S, Barry JL, Cloarec D, Bornet F et al. (1996). Relations between transit time, fermentation products, and hydrogen consuming flora in healthy humans. Gut 38: 870–877.
Fite A, Macfarlane GT, Cummings JH, Hopkins MJ, Kong SC, Furrie E et al. (2004). Identification and quantitation of mucosal and faecal Desulfovibrios using real time polymerase chain reaction. Gut 53: 523–529.
Gagen EJ, Denman SE, Padmanabha J, Zadbuke S, Al Jassim R, Morrison M et al. (2010). Functional gene analysis suggests different acetogen populations in the bovine rumen and tammar wallaby forestomach. Appl Environ Microbiol 76: 7785–7795.
Gibson GR, Macfarlane S, Macfarlane GT . (1993). Metabolic interactions involving sulphate-reducing and methanogenic bacteria in the human large intestine. FEMS Microbiol Ecol 12: 117–125.
Grosskopf R, Janssen PH, Liesack W . (1998). Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl Environ Microbiol 64: 960–969.
Hales BA, Edwards C, Ritchie DA, Hall G, Pickup RW, Saunders JR . (1996). Isolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis. Appl Environ Microbiol 62: 668–675.
Hallam SJ, Girguis PR, Preston CM, Richardson PM, DeLong EF . (2003). Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69: 5483–5491.
Hammer Ø, Harper DAT, Ryan PD . (2001). Past: paleontological statistics software package for education and data analysis]. Palaeont Elec 4: e1–e9.
Huys G, Vanhoutte T, Joossens M, Mahious AS, De Brandt E, Vermeire S et al. (2008). Coamplification of eukaryotic DNA with 16S rRNA gene-based PCR primers: possible consequences for population fingerprinting of complex microbial communities. Curr Microbiol 56: 553–557.
Laue H, Friedrich M, Ruff J, Cook AM . (2001). Dissimilatory sulfite reductase (desulfoviridin) of the taurine-degrading, non-sulfate-reducing bacterium Bilophila wadsworthia RZATAU contains a fused DsrB-DsrD subunit. J Bacteriol 183: 1727–1733.
Leaphart AB, Lovell CR . (2001). Recovery and analysis of formyltetrahydrofolate synthetase gene sequences from natural populations of acetogenic bacteria. Appl Environ Microbiol 67: 1392–1395.
Leloup J, Loy A, Knab NJ, Borowski C, Wagner M, Jorgensen BB . (2007). Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea. Environ Microbiol 9: 131–142.
Levine J, Ellis CJ, Furne JK, Springfield J, Levitt MD . (1998). Fecal hydrogen sulfide production in ulcerative colitis. Am J Gastroenterol 93: 83–87.
Levitt MD, Furne JK, Kuskowski M, Ruddy J . (2006). Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements. Clin Gastroenterol Hepatol 4: 123–129.
Liu WT, Marsh TL, Cheng H, Forney LJ . (1997). Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63: 4516–4522.
Loubinoux J, Bronowicki JP, Pereira IA, Mougenel JL, Le Faou AE . (2002). Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol Ecol 40: 107–112.
Macfarlane GT, Gibson GR, Cummings JH . (1992). Comparison of fermentation reactions in different regions of the human colon. J Appl Bacteriol 72: 57–64.
Mihajlovski A, Alric M, Brugere JF . (2008). A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res Microbiol 159: 516–521.
Miller TL, Wolin MJ . (1985). Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol 141: 116–122.
Miller TL, Wolin MJ, de Macario EC, Macario AJ . (1982). Isolation of Methanobrevibacter smithii from human feces. Appl Environ Microbiol 43: 227–232.
Muyzer G, Teske A, Wirsen CO, Jannasch HW . (1995). Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch Microbiol 164: 165–172.
Nakamura N, Lin HC, McSweeney CS, Mackie RI, Gaskins HR . (2010). Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Ann Rev Food Sci Tech 1: 363–395.
Oxley APA, Lanfranconi MP, Wurdemann D, Ott S, Schreiber S, McGenity TJ et al. (2010). Halophilic archaea in the human intestinal mucosa. Environ Microbiol 12: 2398–2410.
Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB et al. (2010). Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem 285: 22082–22090.
Roediger WE, Moore J, Babidge W . (1997). Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig Dis Sci 42: 1571–1579.
Sahakian AB, Jee SR, Pimentel M . (2010). Methane and the gastrointestinal tract. Dig Dis Sci 55: 2135–2143.
Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R et al. (2007). Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci USA 104: 10643–10648.
Scanlan PD, Shanahan F, Marchesi JR . (2008). Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol 8: 79.
Schmalenberger A, Drake HL, Kusel K . (2007). High unique diversity of sulfate-reducing prokaryotes characterized in a depth gradient in an acidic fen. Environ Microbiol 9: 1317–1328.
Smith CJ, Bryant MP . (1979). Introduction to metabolic activities of intestinal bacteria. Am J Clin Nutr 32: 149–157.
Smith CJ, Danilowicz BS, Clear AK, Costello FJ, Wilson B, Meijer WG . (2005). T-Align, a web-based tool for comparison of multiple terminal restriction fragment length polymorphism profiles. FEMS Microbiol Ecol 54: 375–380.
Springer E, Sachs MS, Woese CR, Boone DR . (1995). Partial gene sequences for the A subunit of methyl-coenzyme M reductase (mcrA) as a phylogenetic tool for the family Methanosarcinaceae. Int J Syst Bacteriol 45: 554–559.
Strocchi A, Furne J, Ellis C, Levitt MD . (1994). Methanogens outcompete sulphate reducing bacteria for H2 in the human colon. Gut 35: 1098–1101.
Tamura K, Dudley J, Nei M, Kumar S . (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Urban E, Hortobagyi A, Szentpali K, Nagy E . (2004). Two intriguing Bilophila wadsworthia cases from Hungary. J Med Microbiol 53: 1167–1169.
Wagner M, Roger AJ, Flax JL, Brusseau GA, Stahl DA . (1998). Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J Bacteriol 180: 2975–2982.
Weaver GA, Krause JA, Miller TL, Wolin MJ . (1986). Incidence of methanogenic bacteria in a sigmoidoscopy population: an association of methanogenic bacteria and diverticulosis. Gut 27: 698–704.
Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y et al. (2009). Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA 106: 2365–2370.
Zinkevich VV, Beech IB . (2000). Screening of sulfate-reducing bacteria in colonoscopy samples from healthy and colitic human gut mucosa. FEMS Microbiol Ecol 34: 147–155.
Zoetendal EG, Heilig HGHJ, Klaassens ES, Booijink CCGM, Kleerebezem M, Smidt H et al. (2006). Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat Protoc 1: 870–873.
Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans ADL, de Vos WM . (2002). Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol 68: 3401–3407.
Acknowledgements
This work was supported by a grant from the Carle Foundation as well as a Carle Hospital-University of Illinois Translational Research Program grant. We thank Justin Ang, David Lee, John Lee and Mark Russell for technical assistance. The subjects are thanked for their participation in the study and Carle Foundation Hospital staff for cooperation in obtaining specimens and subject data. We thank Ann Benefiel for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Nava, G., Carbonero, F., Croix, J. et al. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J 6, 57–70 (2012). https://doi.org/10.1038/ismej.2011.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.90
Keywords
This article is cited by
-
H2 generated by fermentation in the human gut microbiome influences metabolism and competitive fitness of gut butyrate producers
Microbiome (2023)
-
Hydrogen and formate production and utilisation in the rumen and the human colon
Animal Microbiome (2022)
-
Spatial Characteristics of Colonic Mucosa-Associated Gut Microbiota in Humans
Microbial Ecology (2022)
-
Bacteria and Methanogens in the Human Microbiome: a Review of Syntrophic Interactions
Microbial Ecology (2022)
-
Colonisation of the colonic mucus gel layer with butyrogenic and hydrogenotropic bacteria in health and ulcerative colitis
Scientific Reports (2021)
