
Abstract
The spatial heterogeneity of diversity and abundance of diazotrophs were investigated off the Vietnamese coast in the South China Sea (SCS). The study area extended from the Mekong River plume to the shelf region and beyond to stations extending to 1700 m depth. The SCS diazotroph community, based on nifH gene diversity, had components closely related to sequences from open ocean, estuarine, saltmarsh and microbial mat communities. Rarefaction analysis suggested that by using a 97% similarity operational taxonomic unit definition, the majority of nifH sequence diversity in the samples was covered by the 384 nifH clones obtained. The majority of the nifH sequences recovered fell into two clusters: one comprised of Trichodesmium sequences and the other an α-proteobacterial group. Unicellular cyanobacterial groups A and B, and symbiotic filamentous cyanobacterial diazotrophs were detected sporadically. Trichodesmium was by far the most abundant diazotroph, with up to 6 × 105 nifH gene copies per liter. Quantitative PCR probe–primer sets were designed and used to quantify two proteobacterial groups, revealing abundances up to 103–104 nifH gene copies per liter, with the highest abundances in the photic zone. Major components of the clone library were also revealed by a nifH microarray and multidimensional scaling (MDS) analysis. MDS showed that samples from the >10 μm size fraction from 0- to 5-m depths clustered separately from the rest of the samples, primarily due to the abundance of Trichodesmium sequences. The SCS diazotroph community has a relatively low diversity and is a mixture of both estuarine and oceanic fingerprints.
Similar content being viewed by others

Introduction
Several major groups of nitrogen (N2)-fixing microorganisms have been reported in oligotrophic oceans. The filamentous, bloom-forming cyanobacterium Trichodesmium spp., found in tropical oceans around the world, is a major contributor to oceanic N2 fixation and carbon fixation (Capone et al., 1997). Warm, stratified, nitrogen (N)-limited but phosphate (PO43−)- and Fe-replete conditions are associated with Trichodesmium blooms (Sanudo-Wilhelmy et al., 2001; Breitbarth et al., 2007; Webb et al., 2007). Unicellular cyanobacterial diazotrophs were first reported from station ALOHA in the North Pacific Ocean (Zehr et al., 2001) and are now known to have a wide geographic range (Falcón et al., 2002; Langlois et al., 2005; Hewson et al., 2007b). Heterotrophic diazotrophs have also been detected and quantified on the basis of the nifH gene in the open ocean photic zone (Zehr et al., 1998; Falcón et al., 2004; Bird et al., 2005; Church et al., 2005a). In addition, diverse N2-fixing communities have been reported in the deep sea (Mehta et al., 2003), coastal waters (Man-Aharonovich et al., 2007), estuaries (Jenkins et al., 2004) and tropical lagoons (Hewson et al., 2007b). Open ocean diazotroph communities appear to differ from these communities (Zehr et al., 2003). The South China Sea (SCS) is a large marginal sea that potentially shares microbial community components from coastal and open ocean ecosystems. Investigations of nifH diversity and abundances in the SCS may elucidate the factors that control the transition of diazotroph communities from estuarine and coastal to open ocean systems.
The SCS has a deep basin with a maximum depth of 5000 m and a shelf less than 100 m deep. The main water exchange with the Western Pacific Ocean occurs through the Luzon Strait with a sill at the 1900 m depth (Figure 1). The central gyre is permanently stratified and oligotrophic, and N limitation of phytoplankton growth has been reported (Wu et al., 2003), suggesting that the environment is favorable for nitrogen (N2) fixation. Rate measurements suggest that production based on N2 fixation contributes approximately 10% of total production, with the highest rates measured during the monsoon seasons in winter and summer and the lowest rates during intermonsoon (Voss et al., 2006). Bloom-forming filamentous N2-fixing cyanobacterium Trichodesmium spp. and the diazotrophic heterocystous cyanobacterial diatom symbionts are commonly found (Chen et al., 2003, 2004), and a recent study suggested that unicellular diazotrophic cyanobacteria may be present (Chou et al., 2006). Molecular characterization and quantification of the diazotroph community in the SCS have not been reported in the SCS prior to this study.

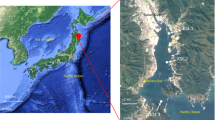
Map of the sampling locations in the study area (Schlitzer, 2007).
Comparative diversity studies are often hampered by the need to create large clone libraries to characterize true sequence diversity (Kemp and Aller, 2004; Hughes and Hellman, 2005). Once the clone libraries are developed, another large effort is required if separate targets are quantified by real-time PCR. Consequently, comparisons of diversity between entire diazotroph communities in samples from different marine systems have largely been prevented by a lack of suitable methodology. Recently, we have developed a nifH oligonucleotide microarray for community analysis of diazotrophs (Moisander et al., 2006, 2007). The microarray is composed of probes targeting cultivated diazotrophs and sequences from uncultivated organisms from various marine environments. The microarray is a useful tool for comparisons of nifH diversity among communities (Hewson et al., 2007a, 2007b).
In this study, we characterized the diversity and abundances of diazotrophs in the SCS during the intermonsoon season using three parallel methods: cloning and sequencing, quantitative PCR, and the nifH microarray. The diversity of nifH genes had not been investigated previously in the SCS, therefore a relatively large clone library was generated. Abundances of the major diazotroph groups were investigated by quantitative PCR using probe and primer sets developed in this and prior studies (Church et al., 2005a, 2005b; Foster et al., 2007). Finally, the use of nifH microarray was evaluated as an alternative method to investigate relative abundance and diversity of nifH genes in the SCS.
Materials and methods
Sampling was conducted during a research cruise of RV Sonne on the Vietnamese coast shelf epipelagic and mesopelagic waters during 12–22 April 2006 (Figure 1). This study occurred during the intermonsoon season, soon after the end of the northeast monsoon. DNA and RNA samples were collected from the water column from depths ranging from surface (0 m) to 1700 m (Supplementary Table S1). Seawater was sampled using Niskin bottles and collected in acid-washed polycarbonate or polyethylene containers. A total of 500–2000 ml seawater was filtered using a peristaltic pump. Samples were passed through 10-μm pore size polyester (GE Osmonics, Trevose, PA, USA) and 0.2-μm membrane (Supor; Pall Gelman, East Hills, NY, USA) filters (25-mm diameter). The filters were placed in sterile cryotubes and snap frozen in liquid nitrogen. The tubes were kept at −80 °C for long-term storage. Additional samples were collected from two experimental studies (mesocosm and microcosm) (M Voss et al., unpublished; PH Moisander et al., unpublished). Briefly, in a 6-day mesocosm experiment, carried out in 600-l high density polyethylene (HDPE) containers on the ship deck, three treatments were included as duplicate: (1) station 32 surface water (control), (2) station 32 surface water mixed with station 32 water from the chlorophyll maximum (upwelling treatment, UW) and (3) station 32 surface water mixed with water from the Mekong River, collected prior to the cruise, and nitrogen silica, and phosphorus added (Mekong River treatment, MR). A microcosm experiment was carried out in 20-l polyethylene containers. Water for the 3-day microcosm experiment was collected at station DS_02, and the treatments included a control and a phosphorus addition.
DNA was extracted using the Qiagen Plant Minikit (Valencia, CA, USA) with modifications. First, 400 μl AP1 buffer (from the kit) was added to the sample tube. The tubes were kept at 65 °C until the filters had thawed and then placed into a dry ice/ethanol bath until frozen solid. The freeze-thaw cycle was repeated three times, and 0.1 g autoclaved glass beads was added. The tubes were agitated for 2 min in a bead beater (Mini-Beadbeater-96; Biospec Inc., Bartlesville, OK, USA), briefly centrifuged and 45 μl Proteinase K (Qiagen; 20 mg ml−1) was added. The tubes were placed in plastic bags and kept at 55 °C with slow rocking motion for 1 h. RNase A (4 μl) was added, and after a brief vortex, the tubes were placed at 65 °C for 10 min. The filters were removed and disposed of using a sterile needle, 120 μl AP2 buffer (from the Qiagen Plant kit) was added, tubes were vortexed and kept in ice for 10 min. Next, the tubes were centrifuged for 5 min at 12 000 g, and the supernatant was added to a QiaShredder column (Qiagen Plant Minikit). The rest of the protocol follows manufacturer's instructions. The final elution volume was 100 μl of AE buffer, with two elutions of 50 μl AE buffer each.
To amplify nifH targets, PCR was carried out as described in Zehr and Turner (2001) (Supplementary Table S1). PCR mix was made with 2.5 mM MgCl2, 1 μM nifH primers nifH3 and nifH4, 200 μM dNTPs and 2 U Taq polymerase (Promega, Madison, WI, USA). Two microliters of DNA extract was added as a template, and the final reaction volume was adjusted to 50 μl with 5 kDa filtered water. The PCR program consisted of an initial 2 min denaturation at 95 °C, then 25 cycles of 30 s at 95 °C, 30 s at 57 °C and 1 min at 72 °C, and final elongation of 7 min at 72 °C. One microliter of template from the first round nested PCR was used in the second round reaction, and primers were replaced with nifH1 and nifH2. The second round PCR consisted of 25 rounds at the same temperatures as the first round. Amplification products of approximately 359 bp were electrophoresed on a 1.2% TAE gel, gel purified (Qiagen Gel Extraction kit) and cloned into a pGEM-t vector (Promega). The plasmid DNA from overnight clones was purified using a 96-well Millipore (Billerica, MA, USA) Montage system. Sequencing was done at the University of California Berkeley sequencing facility. Sequences were manually trimmed and sequence quality checked using the GCG (Accelrys) software package for UNIX. The sequences were aligned in ARB (Ludwig et al., 2004) using a Hidden Markov Model alignment algorithm and a PFAM seed alignment (http://pfam.sanger.ac.uk). Neighbor-joining trees were constructed in ARB with Kimura correction (Kimura, 1983) with translated sequences. For rarefaction analysis, a DNA distance matrix was generated in ARB and imported to DOTUR (Schloss and Handelsman, 2005). The program assigns sequences to operational taxonomic units (OTUs) based on a user-defined sequence similarity threshold. Rarefaction curves were generated using the furthest neighbor method, with OTU threshold values of 100%, 99%, 98% and 97% sequence identity at the DNA level.
Quantitative PCR (qPCR) analysis was carried out as described in Short and Zehr (2005) with slight modifications. Six qPCR probe–primer sets that were previously designed for oceanic cyanobacterial diazotrophs were used (Table 1). In addition, two new quantitative PCR primer–probe sets were designed and used in this study. New primer–probe sets were designed for the sequences 24809A06 (EU052488) and 24774A11 (EU052413). In the nifH phylogenetic tree, 24809A06 clustered with α-Proteobacteria and 24774A11 clustered with γ-Proteobacteria. The primer–probe sets were designed using Primer Express software (Applied Biosystems, Foster City, CA, USA). Cross reactivity of the primer–probe sets was checked against the nifH database with approximately 8000 nifH sequences from GenBank and our unpublished studies. The potential of the new primer–probe sets to amplify mismatch targets was tested using dilution series of plasmids containing inserts matching all of the other primer–probe sets. Additional plasmid preparations from this study that had a range of number of mismatches with the primers and probes (Tables 1 and 2) were also tested. Targets from the DNA extracts from the different size fractions and depths were detected selectively (Supplementary Table S1). Proteobacterial, group A and group B qPCR probes were used to detect target in the 0.2–10 μm size fraction, whereas Trichodesmium probe was used for both 0.2–10 μm and the >10 μm size fractions. Symbiont probes were used for >10 μm size fraction samples at the 0–5 m depth only.
A nifH oligonucleotide microarray was used to characterize diazotroph communities on selected samples (Supplementary Table S1). The samples were chosen to represent coastal and open sea locations, and surface (5 m) and deep samples (several depths below 200 m). From the surface samples, both 0.2–10 μm and >10 μm size fraction was analyzed, if nifH amplification was successful, whereas only the 0.2–10 μm size fraction was analyzed for the deep samples. The microarray was constructed as previously described (Moisander et al., 2007), with slight modifications. Sense-strand 60-mer oligonucleotide probes targeting nifH in a variety of microorganisms were acrydite-modified at the 5′-end (Integrated DNA Technologies, Iowa City, IA, USA). All probes were diluted robotically (Hydra 96; Matrix Technologies, Hudson, NH, USA) to two identical 384-well array printing plates from stock solutions. Ten microliters of probe and 10 μl 2 × Universal EZ rays (Matrix Technologies) spotting buffer with 0.01% Sarkosyl were distributed to each well to a final concentration of 25 μM oligonucleotide per well. The arrays were printed on EZ rays Universal slides using a 16-pin setup in a custom-built microarray printer at the University of California Santa Cruz and ArrayMaker software (Joseph DeRisi, University of California San Francisco). After spotting, slides were stored at room temperature in the dark. The slides were treated according to the manufacturer's instructions (EZ rays Universal slide kit; Matrix Technologies) prior to and after the printing. To evaluate delivery of probes to the slides, OliGreen (Molecular Probes, Eugene, OR, USA) staining in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) was used. The array was stained for 30 min under a coverslip, rinsed twice with TE buffer for 10 s, then dried in a filtered air stream and scanned at 532 nm with a GenePix 4000B scanner (Axon Instruments, Sunnyvale, CA, USA). The staining indicated that all pins delivered probe to the slides until the end of the run. The temperature and relative humidity during the print run averaged 22–23 °C and 21–22%, respectively. A total of 706 nifH probes and five control probes were included on the array. Each nifH probe was printed four times on each slide (twice from two print plates each), resulting in a total of 3072 nifH features on each slide. On each array slide, each of the five control probes was printed 64 times, and 448 additional features had buffer only. The total number of features on each slide was 3840. The volume was checked on several random wells on print plates at the end of the run, and evaporation during the print run was shown to be insignificant.
Targets for microarray hybridizations were amplified using the nested nifH primer set (Zehr and Turner, 2001). The PCR mix was made with 4 mM MgCl2, 1 μM nifH primers, 200 μM dNTPs and 2 U Taq polymerase (Go-Taq; Promega) (final concentrations). Two microliters of DNA extract was added as template and the final reaction volume was adjusted to 50 μl with 5 kDa filtered water. The PCR program consisted of an initial 2-min denaturation at 95 °C, then 30 cycles of 30 s at 95 °C, 30 s at 57 °C and 1 min at 72 °C, and final elongation step of 7 min at 72 °C. One microliter of template from the first round nested PCR was used in the second round reaction, and primers were replaced with nifH1 and nifH2. The second round PCR consisted of 25 rounds at the same temperatures as the first round. Amplification products were electrophoresed on a 1.2% TAE gel and gel-purified (Qiagen Gel Extraction kit). The PCR product was quantified using the PicoGreen assay (Molecular Probes) and adjusted to 5 ng μl−1 prior to biotinylation, unless otherwise specified. Biotinylation was carried out using a BrightStar biotinylation kit (Ambion, Austin, TX, USA). Ten microliters of PCR product, 5 μl positive control PCR mix and 5 μl TE buffer were combined. The control mix composition, biotinylation, hybridization, secondary staining (using streptavidin-conjugated Alexa-555) and washing steps followed protocols described previously (Moisander et al., 2006, 2007). Each sample was hybridized on two microarray slides, processed on different days.
Microarray fingerprints were analyzed using multidimensional scaling (MDS) (SPSS v. 12.0 for Windows). To minimize the effects of potential cross hybridization, only probes that had an identity of 90% or less among the probe set (706 probes, total) were included, resulting in 479 probes in the analysis (Moisander et al., 2007). Signal that was less than 7% of the maximum signal in the hybridization was considered background and was excluded (Moisander et al., 2006). Finally, the signal intensity data from each probe were converted to proportions of the total signal intensity in the array. GenBank submission numbers for the sequences from this study are EU052295–EU052680.
Results
A total of 384 clones were recovered from the samples, representing 11 stations. On the basis of phylogenetic analysis, nifH sequences from the samples fell in nifH Clusters 2 and 3, following cluster definitions by Zehr et al. (2003) (Figures 2 and 3). Cluster 2 includes cyanobacteria, α-, β- and γ-Proteobacteria, whereas Cluster 3 includes many anaerobes, including δ-Proteobacteria. In cyanobacteria, sequences originating from Trichodesmium spp. formed a major cluster (95% amino-acid sequence identity), with a total of 156 sequences (Figure 2a, Clade 1). Trichodesmium spp. sequences made up the highest number of any single sequence type in the clone library. Trichodesmium sequences were obtained from the photic zone to a depth of 75 m, with most of the sequences recovered from the surface samples (0–5 m depth). At station 84, a sequence was recovered from 75 m that clustered with the recently discovered oceanic unicellular cyanobacterium (group A) (Figure 2a, Clade 2) (Zehr et al., 2001). Group A sequence had a 100% protein identity with sequences previously recovered from North Pacific Ocean (numerous sequences from various studies), Chesapeake Bay (Short and Zehr, 2007) and Atlantic Ocean (Langlois et al., 2005). One cyanobacterial sequence was recovered from an enrichment incubation (microcosm experiment) from the Mekong plume water and had 98% identity with the Cyanothece sp. ATCC 51142 nifH sequence and 100% protein identity with sequences from open ocean (Langlois et al., 2005) but only 86% identity with the sequence from Crocosphaera watsonii.

Phylogenetic trees for nifH constructed using the neighbor-joining method for (a) cyanobacterial and (b) Cluster 3 sequences. Numbers (1B, 1B and so on) refer to groupings by Zehr et al. (2003) based on DNA and amino-acid sequences, respectively. The number of sequences included in a collapsed clade is shown.

Phylogenetic trees for nifH constructed using the neighbor-joining method. The sequences cluster with (a) α-, β- and (b) γ-proteobacterial sequences. Numbers (1J2, 1J and so on) refer to groupings by Zehr et al. (2003) based on DNA and amino-acid sequences, respectively. The number of sequences included in collapsed clades is shown.
Seven sequences belonging to the nifH Cluster 3 were found in the clone library (Figure 2b). These sequences were very similar to sequences previously recovered from various environments (saltmarshes, sediments, Chesapeake Bay water column, coral reefs, microbial mats). None of the Cluster 3 sequences from this study had close cultivated relatives.
A large group of sequences fell in a cluster composed of α-Proteobacteria (Figure 3a) (153 sequences, Clade 1, 92% identity among sequences). The dominant sequence type in this clade was very similar to a previously sequenced group of reverse transcription PCR clones from the North Pacific (Falcón et al., 2004), and one of these sequences was included as a probe in the nifH microarray (AY191947). AY191947 has a 99% DNA sequence identity with the SCS sequence from this cluster that was used to design a qPCR probe and primer set. In this study, the α-proteobacterial sequence was recovered in both surface and deep samples (Figure 3a). A representative dominant sequence type from this clade (EU052488 from the cluster of 153 sequences) had a 95% protein sequence identity with the nifH sequence from the α-Proteobacterium Sinorhizobium meliloti, and a 97% identity with microbial mat and coral reef sequences. Two other distinct sequence types had several repeats in the cluster with α- and β-Proteobacteria. Clade 2 (Figure 3a) was a group of seven sequences from this study, with 95% or higher protein identity. A dominant sequence type from this cluster (EU052346) had a 97% protein identity with the α-Proteobacterium Sphingomonas azotifigens and with uncultivated sequences from a mangrove system and English Channel. Clade 3 (95% protein identity or higher) included sequences that were 100% identical to the Burkholderia vietnamiensis nifH sequence.
Several repeats of proteobacterial nifH amino-acid sequences were obtained from 5–68 m depths that formed a distinct cluster, with 92% identity (Figure 3b, Clade 1). A dominant representative sequence from this cluster (EU052537) had a 94% identity with the Geobacter metallireducens nifH sequence and 98–99% protein identity with many environmental sequences including ones previously obtained from Lake Michigan, Chesapeake Bay, mangroves in China and the Mediterranean Sea. A group of 19 sequences was recovered that formed a clade with 95% protein identity (Figure 3b, Clade 2). A dominant sequence type from this cluster (EU052409) had a 98% protein sequence identity with the γ-proteobacterial nifH sequence that was expressed at the North Pacific Ocean HOT time-series station ALOHA (DQ269145; Church et al., 2005b) and 99% identity with a sequence expressed in the Arabian Sea (AY800142; Bird et al., 2005). A probe (DQ269145) targeting this sequence was included in the microarray. Several other sequences were recovered that clustered with sequences previously obtained from marine microbial mats, estuaries and wetlands.
Rarefaction analysis was carried out to estimate how well the sampling effort covered the real nifH diversity in the SCS. A rarefaction curve was generated using four threshold values for OTU; 100%, 99%, 98% and 97% (Figure 4). If each unique sequence was considered a different OTU, the rarefaction curve showed an increasing slope and suggested that the sampling effort did not cover all of the diversity in the system. If 97–98% identity was used as a threshold to discriminate an OTU, the curve appeared to be reaching a plateau.

Rarefaction analysis for the nifH sequence data. The curves were generated for four levels of operational taxonomic unit (100%, ⩾99%, ⩾98% or ⩾97% identity).
Diazotroph community composition was analyzed by a microarray in the surface layer at selected stations (0.2–10 μm size fraction at stations 56, 72 and 76; >10 μm size fraction at stations 42, 56, 76, 72, 84 and a mesocosm sample) and deep water samples from several depths below 200 m at stations 44, 56 and 66 (0.2–10 μm size fraction) (Figures 1, 5, 6). The microarray fingerprints paralleled the clone library data. In all >10 μm-size fraction samples except the mesocosm sample, the highest microarray signal was present in the probes targeting Trichodesmium (Figure 5). The only additional signal that was present in several samples was from several α-proteobacterial probes, including probes for uncultivated sequences and a Rhodobacter sp. sequence. In microarray hybridizations with the nifH amplified from the 0.2–10 μm size fraction from the surface 0–5 m depth, the α-proteobacterial (probe AY191947) signal was high but other probes detected target as well. Azospirillum brazilense, a microbial sequence from a stromatolite, and AF389819 were detected with high signal intensities, and a probe targeting the γ-proteobacterial sequence DQ269145 was detected at all of the stations in the surface small-size fraction samples. Trichodesmium and group A were also detected at low signal intensities in three and two of the small-size fraction surface samples, respectively. The probe AY224030 for a Chesapeake Bay sequence produced an exceptionally high signal at station 76.

Typical microarray fingerprints (normalized fluorescence units) from (a) station 72, 5 m depth, in the 0.2–10 μm size fraction, (b) station 42, 5 m depth, >10 μm size fraction and (c) station 66, 1500 m depth, 0.2–10 μm size fraction. Microarray probes (479 probes total) are grouped based on phylogenetic affiliation (Zehr et al., 2003, Moisander et al., 2007). α, α-Proteobacteria; A, Archaea; β, β-Proteobacteria; C1, Cluster 1; C3, Cluster 3; Cy, Cyanobacteria; δ, δ-Proteobacteria; F, Firmicutes; γ, γ-Proteobacteria; u, unclassified.

Multidimensional scaling analysis for microarray fingerprints (stress=0.044). Normalized data averaged from replicate array hybridizations of each sample are shown. The signal intensity data from each sample were converted to proportion of total signal intensity in each sample. Only microarray probes that had <90% identity were included in the analysis. The samples are coded as follows: 76_S0.2u, station 76, surface 0–5 m, 0.2–10 μm size fraction; 66_800 m, station 66, 800 m depth, and so on. All of the samples below 210 m were determined from the 0.2–10 μm size fraction. Clusters for >10 μm size fraction surface samples and mesopelagic samples are circled with dotted lines.
Vertical profiles of the diazotroph communities were analyzed by the nifH microarray on samples from deep water (0.2–10 μm size fraction) at stations 44, 56 and 66. Microarray fingerprints from all deep stations 44 (from 210 to 840 m), 56 (from 400 to 1700 m) and 66 (from 400 to 1500 m) were very similar (station 66 shown in Figure 5). The microarray signal in mesopelagic samples was mostly from the same α-proteobacterial probes that had positive signal in the surface 0.2–10 μm samples. Several probes that detected target in surface waters, including Trichodesmium, did not detect target in the mesopelagic samples. A few additional targets were detected at lower signal intensities and less consistently among samples.
To carry out an MDS analysis for the microarray data, the signal above background from each probe was transformed to proportion of total signal intensity in the hybridization. Data from all microarray hybridizations were included in the same MDS analysis. The MDS with all data included produced a low stress value (0.044). MDS plots with stress values <0.1 are considered to give good ordinations with little risk for misinterpretation (Clarke et al., 1993; Kan et al., 2006). The data clustered as expected based on visual observations of the array hybridization fingerprints. The samples from >10 μm size fraction (surface layer only) clustered as a unique group, with the exception of the sample from station 72 and a sample from the mesocosm experiment. The two size fractions from station 72 had very similar microarray fingerprints, and the >10 μm sample from the mesocosm clustered with the mesopelagic samples. Communities in samples from the 0.2–10 μm size fraction from surface 0–5 m were distinct from the mesopelagic communities (200–1700 m) on the first dimension, and station 76 community in the 0.2–10 μm size fraction was distinct from communities in all other samples on the second dimension.
Quantitative PCR was used to enumerate nifH gene abundances of the most abundant diazotrophs. The data from cross-reactivity tests showed that the new α- and γ-proteobacterial primer–probe sets (24809A06 and 24774A11, respectively) did not detect any of the dominant ocean diazotrophs (group A, group B, Trichodesmium and three symbiont cyanobacteria) (Table 2). Low cross reactivity was detected between the α- and γ-proteobacterial probes. However, the difference in amplification efficiency was three to four orders of magnitude and therefore would not significantly bias the data. QPCR data and microarray signal intensities were compared (Figure 7). Data were pooled from all samples that had data from both methods, resulting in 10 (α-Proteobacterium), 11 (γ-Proteobacterium) or 19 (Trichodesmium spp.) samples. There was a positive, although at times a weak, relationship between nifH gene copies detected by qPCR and microarray signal intensities (Figure 7). The slope of the relationship appeared to vary between probes. These relationships were not tested statistically because the data did not satisfy normality and homoscedasticity even after transformations.

Comparison of nifH gene abundance (genes per liter) determined by qPCR and signal intensity from nifH microarray hybridizations.
Trichodesmium was detected in the surface layers throughout the study area and had by far the highest nifH gene abundances of any diazotroph detected by qPCR, with maximum abundances of 6 × 105 nifH copies per liter (Figure 8). Highest nifH abundances were detected between 5 and 40 m depths, with peaks at stations 74 and 84, and slightly lower but similar abundances at stations M13, 56, 83 and 76 (Figure 9, Supplementary Table S1). Unicellular group A cyanobacteria, α-proteobacterial group 24809A06 and symbionts had the maximum nifH gene abundances in the order of 104 per liter (Figure 8). The α-proteobacterial qPCR probe 24809A06 detected targets from the euphotic layers to mesopelagic depths. The γ-Proteobacterium 24774A11 and group B cyanobacteria were detected in the euphotic layers with up to 103 nifH genes per liter.

Depth distributions of diazotroph nifH abundances (genes per liter) determined by quantitative PCR. In symbionts: het-1, circles; het-2, triangles.

Maximum nifH gene abundance (per liter) of Trichodesmium in the study area at each station. Color of the dots indicates the nifH abundance at the depth in which the abundance was the highest. There was no data from the station indicated with black.
Discussion
Dissolved inorganic nitrogen is replenished in the SCS to subsurface layers in summer during southwest monsoon through updwelling, coinciding with decreased mixed layer depth; however, there is year-to-year variability (Chou et al., 2006; Dippner et al., 2007). The seasonal cycle in N2 fixation appears to be linked to this climatically driven seasonality in hydrodynamics and production in the system. Previously, it was estimated that N2 fixation provides up to 10% of the total carbon production in the SCS (Voss et al., 2006; Loick et al., 2007). The highest potential for N2 fixation was suggested to occur during the summer monsoon with no contribution during spring intermonsoon (Loick et al., 2007). In contrast, in this study that also occurred during the spring intermonsoon, Trichodesmium was present throughout the study area and its nifH genes were found at high abundances. According to this study, other groups of diazotrophs are also present in the SCS through the intermonsoon, potentially contributing to N2 fixation.
Abundances of clones in the clone library, microarray signal intensity and quantification by quantitative PCR paralleled each other for Trichodesmium abundance in the study area. This globally important cyanobacterium was clearly a dominant diazotroph in the SCS during the study period. In previous studies in the SCS, Trichodesmium abundances were estimated to be up to 48–280 trichomes per liter (Chen et al., 2003; Wu et al., 2003). Assuming that one trichome consists of approximately 100 cells (Capone et al., 1997) and that each cell contains one genome, the maximum Trichodesmium abundances in this study were approximately 2 × 103–6 × 103 trichomes per liter, suggesting that abundances were about one order of magnitude higher than previously reported from the SCS. The nifH gene abundances in this study were in a similar range with numbers reported from station ALOHA in the North Pacific (Church et al., 2005a, 2005b; Zehr et al., 2007) and the Amazon River plume (Foster et al., 2007). The Trichodesmium genome has one nifH copy, but if one cell has replicate genomes, the nifH gene abundances would overestimate cell abundances. Currently, little is known about the potential of multiple genomes in cells of Trichodesmium or other cyanobacteria. Trichodesmium abundances had an increasing trend with distance from the Mekong River plume, potentially reflecting sources of nutrients maintaining the cyanobacterial biomass. On the basis of the salinity data in the study area, Mekong plume quickly turned southwards with the prevailing currents and had a very limited influence in the study area (M Voss et al., unpublished data). Trichodesmium abundances in the SCS have been proposed to reflect intrusions of the Kuroshio current (Chen et al., 2003), and their low abundance and low N2-fixation rates in the system were previously suggested to be controlled by Fe limitation (Wu et al., 2003).
The abundant α-proteobacterial sequence was very similar to a sequence from the North Pacific Ocean that expressed nifH both during the day and during the night (Falcón et al., 2004). A probe was included on the microarray that targeted this group of sequences, and a quantitative PCR probe (24809A06) was designed and applied to characterize its abundance and distribution. The results from the different approaches paralleled each other but also produced somewhat different results. The sequence was recovered by cloning numerous times from the mesocosm water sample and several times from surface and deep water samples at various stations, and detected by qPCR at six stations of the 22 stations sampled. The maximum nifH gene abundances for the α-Proteobacterium were 6 × 104 nifH copies per liter in the epipelagic and 7 × 102 in the mesopelagic waters. This probe detected target in all of the microarray hybridizations, with extremely high signals in the mesocosm water and at stations 72 and 56 in the 0.2–10 μm size fraction (40, 18 and 26 fluorescence units, respectively). In contrast, at station 76, the α-Proteobacterium was not detected by qPCR, and microarray signal was much lower (3.2 fluorescence units) than that at other stations. These results suggest that the nifH microarray may have a better capacity to detect presence of low-abundance targets than qPCR. It is possible that the α-proteobacterial sequence has a relatively higher amplification efficiency compared to other targets, especially in mesopelagic samples where other targets may have been rare. Consequently, its abundance may be overestimated based on the number of clones recovered and signal intensities in the microarray. In addition, a large proportion of the α-proteobacterial sequences were recovered from the mesocosms in which conditions may have selectively enriched this microbe. In spite of the frequent observations of this diazotroph from the open ocean, the ecology of this microorganism is as elusive as that of most uncultivated microbes. In this study, we detected it at high abundances in enrichment cultures (mesocosm) but it was also present consistently in surface waters and frequently in mesopelagic waters. The qPCR primer–probe set developed in this study will be useful in future studies characterizing its distribution and expression patterns. Four sequences that had a 100% match with B. vietnamiensis were obtained in the water from the mesocosm. Although presence of B. vietnamiensis could indicate contamination, B. vietnamiensis is also present in reservoirs and agricultural settings where it actively fixes N2 (Menard et al., 2007). The dominant α-Proteobacterium was relatively distant from B. vietnamiensis, with a 90% protein identity.
A γ-Proteobacterium was present with multiple repeats in the clone library that was very similar to an nifH gene sequence expressed in previous studies (Church et al., 2005b). Quantitative PCR showed that this microbe was frequently detected in the upper 50-m surface layers but not deeper in the water column. This group of bacteria was also expressing nifH in the surface waters in the Arabian Sea (Bird et al., 2005). The vertical distribution suggests that it may benefit from light, potentially using bacteriochlorophyll or a rhodopsin-based metabolism.
Almost 400 clones were obtained in this study, representing a wide range of depths and stations in the study area. The two most abundant sequence types, Trichodesmium spp. and the α-Proteobacterium AY191947 formed more than ¾ of the entire clone library, and the rest fell under proteobacterial, cyanobacterial and ‘Cluster 3’ sequences. Rarefaction analysis was carried out to estimate whether the clone library was large enough to represent the true nifH diversity in the samples from the study area. Using the 16S rRNA gene sequence, it was previously estimated that approximately 690 clones were necessary to capture the true microbial diversity from the Sargasso Sea, whereas more than 10 000 clones were necessary from an agricultural soil (Schloss and Handelsman, 2005). The rarefaction curves in this study suggest that if 97% identity is considered an OTU, generally defined as bacterial ‘species’ based on 16S rRNA genes, the majority of the nifH sequence diversity was captured with the 384 clones. However, if each unique DNA sequence is considered to be an OTU, the cloning effort was not nearly sufficient. It appears that the sequence diversity in the system was relatively low compared to most estuarine and benthic systems, but the community included components that reflected open ocean, benthic and estuarine origins. Open ocean photic layer microbial communities appear to have lower nifH diversity and different community composition (Zehr et al., 1998) from coastal and estuarine (Jenkins et al., 2004), and benthic communities, including microbial mats (Omoregie et al., 2005), coral reefs (Hewson et al., 2007b) and salt marshes (Lovell et al., 2001; Moisander et al., 2005). However, in the high-diversity coastal and benthic systems, a large proportion of the community nifH genes is not expressed, at least not at detectable levels (Moisander et al., 2006; Man-Aharonovich et al., 2007; Short and Zehr, 2007). Major oceanic diazotrophs were detected in the SCS, but additional sequences were closely identical with sequences previously obtained from estuarine, coastal and benthic systems, such as salt marshes, Chesapeake Bay and Neuse River estuaries, Monterey Bay, Heron Reef in Australia and microbial mats in Baja California. In many cases, there was high similarity to sequences from previously sequenced environmental samples, such as the group A sequence and many proteobacterial sequences. Surprisingly, the mesopelagic community included γ-Proteobacteria that clustered with many estuarine sequences. Previously, higher diversity than in this study was detected in mesopelagic samples in the North Atlantic and Pacific Oceans (Hewson et al., 2007a).
The presence of unicellular cyanobacteria in the SCS has been proposed before (Chou et al., 2006). Unicellular group B-type cyanobacteria and the uncultivated group A bacterial groups were detected several times in the study area, but their presence was sporadic. It is notable that these diazotrophs were detected during the intermonsoon period when N2 fixation is thought to play the smallest role in the nutrient inputs over the seasonal cycle (Loick et al., 2007). Detection and identification of the unicellular cyanobacteria in the system contribute to our understanding of factors controlling their global distributions.
Overall, the microarray fingerprints revealed the major components that were found in the clone library. The microarray fingerprints were less complex than in a recent microarray study carried out in the Chesapeake Bay (Moisander et al., 2007), reflecting the lower nifH diversity in the SCS. Microarray signal intensities in this study had a positive relationship with data from quantitative PCR; however, the relationship was at times nonlinear and the slope of this relationship was not constant. The nifH microarray approach used is based on PCR amplification, but this approach does not have the cloning bias. The array signal intensity is influenced by other PCR biases such as preferential amplification of certain targets (Suzuki et al., 1998) and Taq fidelity (Barnes, 1992) as well as specificity of the probes. These biases may change with variable composition of nifH targets or DNA extract purity just like in any PCR-based application. Considering all the potential factors that may cause differences between the microarray and qPCR from environmental samples, it is encouraging to see the overall positive relationship between the data sets from the two. The results suggest that nifH microarray signal intensities can be used to detect large changes in abundances of specific diazotrophs in water samples from the same study area. The array is most informative when applied in studies investigating relative changes in community composition. Small changes in individual peak heights should be interpreted with caution and, if possible, verified by qPCR.
This is the first study describing molecular nifH diversity in the SCS. We used several quantitative PCR probes designed in previous studies based on sequences recovered from other oceanic regions and applied an nifH microarray designed using sequences available in 2005, with no probes targeting sequences originating from the study area. The data showed that the microarray captured the major components of the diazotroph community even in this previously uncharacterized ocean region and suggest that we may be at the point of reaching saturation of oceanic nifH diversity in public databases.
References
Barnes WM . (1992). The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene 112: 29–35.
Bird C, Martinez JM, O'Donnell AG, Wyman M . (2005). Spatial distribution and transcriptional activity of an uncultured clade of planktonic diazotrophic γ-proteobacteria in the Arabian Sea. Appl Environ Microbiol 71: 2079–2085.
Breitbarth E, Oschlies A, LaRoche J . (2007). Physiological constrains on the global distribution of Trichodesmium—effect of temperature on diazotrophy. Biogeosciences 4: 53–61.
Capone DG, Zehr JP, Paerl HW, Bergman B, Carpenter EJ . (1997). Trichodesmium, a globally significant marine cyanobacterium. Science 276: 1221–1229.
Chen Y, Chen H-Y, Lin Y-H . (2003). Distribution and downward flux of Trichodesmium in the South China Sea as influenced by the transport from the Kuroshio current. Mar Ecol Prog Ser 259: 47–57.
Chen YL, Chen H-Y, Karl DM, Takahashi M . (2004). Nitrogen modulates phytoplankton growth in spring in the South China Sea. Cont Shelf Res 24: 527–541.
Chou W-C, Chen Y-LL, Sheu DD, Shih Y-Y, Han C-A, Cho CL et al. (2006). Estimated net community production during the summertime at the SEATS time-series study site, northern South China Sea: Implications for nitrogen fixation. Geophys Res Lett 33: L22610.
Church MJ, Jenkins BD, Karl DM, Zehr JP . (2005a). Vertical distributions of nitrogen-fixing phylotypes at Stn ALOHA in the oligotrophic North Pacific Ocean. Aquat Microb Ecol 38: 3–14.
Church MJ, Short CM, Jenkins BD, Karl DM, Zehr JP . (2005b). Temporal patterns of nitrogenase gene (nifH) expression in the oligotrophic North Pacific Ocean. Appl Environ Microbiol 71: 5362–5370.
Clarke KR . (1993). Nonparametric multivariate analyses of changes in community structure. Austra J Ecol 18: 117–143.
Dippner JW, Nguyen KV, Hein H, Ohde T, Loick N . (2007). Monsoon-induced upwelling off the Vietnamese coast. Ocean Dynamics 57: 46–62.
Falcón LI, Carpenter EJ, Cipriano F, Bergman B, Capone DG . (2004). N-2 fixation by unicellular bacterioplankton from the Atlantic and Pacific Oceans: Phylogeny and in situ rates. Appl Environ Microbiol 70: 765–770.
Falcón LI, Cipriano F, Chistoserdov AY, Carpenter EJ . (2002). Diversity of diazotrophic unicellular cyanobacteria in the tropical North Atlantic Ocean. Appl Environ Microbiol 68: 5760–5764.
Foster RA, Subramaniam A, Mahaffey C, Carpenter EJ, Capone DG, Zehr JP . (2007). Influence of the Amazon River plume on distributions of free-living and symbiotic cyanobacteria in the western tropical North Atlantic Ocean. Limnol Oceanogr 52: 517–532.
Hewson I, Moisander PH, Achilles KA, Carlson CA, Jenkins BD, Mondragon EA et al. (2007a). Characteristics of diazotrophs in epi-to abyssopelagic waters of the Sargasso Sea. Aquat Microb Ecol 46: 15–30.
Hewson I, Moisander PH, Morrison AE, Zehr JP . (2007b). Diazotrophic bacterioplankton in a coral reef lagoon: Phylogeny, diel expression, and response to phosphate enrichment. ISME J 1: 78–91.
Hughes JB, Hellman JJ . (2005). The application of rarefaction techniques to molecular inventories of microbial diversity. Methods Enzymol 397: 292–304.
Jenkins BD, Steward GF, Short SM, Ward BB, Zehr JP . (2004). Fingerprinting diazotroph communities in the Chesapeake Bay by using a DNA macroarray. Appl Environ Microbiol 70: 1767–1776.
Kan J, Crump BC, Wang K, Chen F . (2006). Bacterioplankton community in Chesapeake Bay: predictable or random assemblages. Limnol Oceanogr 51: 2157–2169.
Kemp PF, Aller JY . (2004). Estimating prokaryotic diversity: When are 16S rDNA libraries large enough? Limnol Oceanogr Methods 2: 114–125.
Kimura M . (1983). The Neutral Theory of Molecular Evolution. Cambridge University Press: Cambridge, New York, pp 367.
Langlois RJ, LaRoche J, Raab PA . (2005). Diazotrophic diversity and distribution in the tropical and subtropical Atlantic Ocean. Appl Environ Microbiol 71: 7910.
Loick N, Dippner J, Nhu Doan H, Liskow I, Voss M . (2007). Pelagic nitrogen dynamics in the Vietnamese upwelling area according to stable nitrogen and carbon isotope data. Deep Sea Res Part I 54: 596–607.
Lovell CR, Friez MJ, Longshore JW, Bagwell CE . (2001). Recovery and phylogenetic analysis of nifH sequences from diazotrophic bacteria associated with dead aboveground biomass of Spartina alterniflora. Appl Environ Microbiol 67: 5308–5314.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Man-Aharonovich D, Kress N, Bar Zeev E, Berman-Frank I, Beja O . (2007). Molecular ecology of nifH genes and transcripts in the eastern Mediterranean Sea. Environ Microbiol 9: 2354–2363.
Mehta MP, Butterfield DA, Baross JA . (2003). Phylogenetic diversity if nitrogenase (nifH) genes in deep-sea and hydrothermal vent environments of the Juan de Fuca ridge. Appl Environ Microbiol 69: 960–970.
Menard A, Monnez C, de los Santos PE, Segonds C, Caballero-Mellado J, LiPuma JJ et al. (2007). Selection of nitrogen-fixing deficient Burkholderia vietnamiensis strains by cystic fibrosis patients: involvement of nif gene deletions and auxotrophic mutations. Environ Microbiol 9: 1176–1185.
Moisander PH, Morrison AE, Ward BB, Jenkins BD, Zehr JP . (2007). Spatial-temporal variability in diazotroph assemblages in Chesapeake Bay using an oligonucleotide nifH microarray. Environ Microbiol 9: 1823–1835.
Moisander PH, Piehler MF, Paerl HW . (2005). Diversity and activity of epiphytic nitrogen-fixers on standing dead stems of the salt marsh grass Spartina alterniflora. Aquat Microb Ecol 39: 271–279.
Moisander PH, Shiue L, Steward G, Jenkins BD, Bebout BM, Zehr JP . (2006). Application of a nifH oligonucleotide microarray for profiling diversity of N2-fixing microorganisms in marine microbial mats. Environ Microbiol 8: 1721–1735.
Omoregie EO, Crumbliss LL, Bebout BM, Zehr JP . (2005). Comparison of diazotroph community structure in Lyngbya sp and Microcoleus chthonoplastes dominated microbial mats from Guerrero Negro, Baja, Mexico. FEMS Microbiol Ecol 47: 305–318.
Sanudo-Wilhelmy SA, Kustka AB, Gobler CJ, Hutchins DA, Yang M, Lwiza K et al. (2001). Phosphorus limitation of nitrogen fixation by Trichodesmium in the central Atlantic Ocean. Nature 411: 66–69.
Schlitzer R . (2007). Ocean data view. http://odv.awi.de/.
Schloss PD, Handelsman J . (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501–1506.
Short SM, Zehr JP . (2005). Quantitative analysis of nifH genes and transcripts from aquatic environments. Methods Enzymol 397: 380–394.
Short SM, Zehr JP . (2007). Nitrogenase gene expression in the Chesapeake Bay Estuary. Environ Microbiol 9: 1591–1596.
Suzuki M, Rappe MS, Giovannoni SJ . (1998). Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl Environ Microbiol 64: 4522–4529.
Voss M, Bombar D, Natalie L, Dippner JW . (2006). Riverine influence on nitrogen fixation in the upwelling region off Vietnam, South China Sea. Geophys Res Lett 33: L07604. 1–4.
Webb EA, Jakuba RW, Moffett JW, Dyhrman ST . (2007). Molecular assessment of phosphorus and iron physiology in Trichodesmium populations from the western Central and western South Atlantic. Limnol Oceanogr 52: 2221–2232.
Wu J, Chung S-W, Wen L-S, Liu K-K, Chen Y, Chen H-Y et al. (2003). Dissolved inorganic phosphorus, dissolved iron, and Trichodesmium in the oligotrophic South China Sea. Global Biogeochemical Cycles 17: 1008.
Zehr JP, Jenkins BD, Short SM, Steward GF . (2003). Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol 5: 539–554.
Zehr JP, Mellon MT, Zani S . (1998). New nitrogen-fixing micro-organisms detected in oligotrophic oceans by amplification of nitrogenase (nifH) genes. Appl Environ Microbiol 64: 3444–3450.
Zehr JP, Turner PJ . (2001). Nitrogen fixation: Nitrogenase genes and gene expression. In: Paul JH (ed). Methods in Microbiology: Marine Microbiology. Academic Press: London, UK, pp 271–286.
Zehr JP, Waterbury JB, Turner PJ, Montoya JP, Omoregie E, Steward GF et al. (2001). Unicellular cyanobacteria fix N2 in the subtropical North Pacific Ocean. Nature 412: 635–638.
Zehr JP, Montoya JP, Jenkins BD, Hewson I, Mondragon E, Short CM et al. (2007). Experiments linking nitrogenase gene expression to nitrogen fixation in the North Pacific subtropical gyre. Limnol Oceanogr 52: 169–183.
Acknowledgements
We thank Nguyen NL and Nhu DH for providing facilities and assistance at the Nha Trang Oceanographic center, Vietnam. J Montoya, A Subramaniam and D Bombar are acknowledged for discussions. AE Morrison, E Mondragon, R Nguyen and the crew onboard RV Sonne are acknowledged for technical assistance. Funding was provided by NSF OCE-0425363 (JPZ), the Gordon and Betty Moore Foundation (JPZ), German Federal Ministry of Education and Research (Grant no. 03G187A) (MV) and German Science foundation (Grant no. BO768/7-3) (MV).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary information
Rights and permissions
About this article
Cite this article
Moisander, P., Beinart, R., Voss, M. et al. Diversity and abundance of diazotrophic microorganisms in the South China Sea during intermonsoon. ISME J 2, 954–967 (2008). https://doi.org/10.1038/ismej.2008.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.51
Keywords
This article is cited by
-
Diazotrophs are overlooked contributors to carbon and nitrogen export to the deep ocean
The ISME Journal (2023)
-
Cell-specific measurements show nitrogen fixation by particle-attached putative non-cyanobacterial diazotrophs in the North Pacific Subtropical Gyre
Nature Communications (2022)
-
Transcriptional responses of Trichodesmium to natural inverse gradients of Fe and P availability
The ISME Journal (2022)
-
Evidence of the Significant Contribution of Heterotrophic Diazotrophs to Nitrogen Fixation in the Eastern Indian Ocean During Pre-Southwest Monsoon Period
Ecosystems (2022)
-
Activity and distribution of diazotrophic communities across the Cape Verde Frontal Zone in the Northeast Atlantic Ocean
Biogeochemistry (2022)
