
Abstract
Redox sensor green (RSG), a novel fluorescent dye from Invitrogen was employed as a tool for real-time detection of microbes metabolically active in situ, in combination with flow cytometry and cell sorting. Lake Washington sediment, an environment known for high rates of methane oxidation, was used as a model, and methylotrophs were targeted as a functional group. We first tested and optimized the performance of the dye with pure methylotroph cultures. Most cells in actively growing cultures were positive for staining, whereas in starved cultures, few cells fluoresced. However, starved cells could be activated by addition of substrate. High numbers of fluorescing cells were observed in a Lake Washington sediment sample, and activation of subpopulations of cells was demonstrated in response to methane, methanol, methylamine and formaldehyde. The fraction of the population activated by methane was investigated in more detail, by phylogenetic profiling. This approach showed that the major responding species were the Methylomonas species, previously isolated from the site, and Methylobacter species that have not yet been cultivated from Lake Washington. In addition, from the methane-stimulated fraction, uncultivated bacterial sequences were obtained that belonged to unclassified Deltaproteobacteria, unclassified Verrucomicrobiles and unclassified Acidobacteria, suggesting that these microbes may also be involved in methane metabolism. The approach was further tested for its utility in facilitating enrichment for functional types that possess specific metabolic activities but resist cultivation. It was demonstrated that in enrichment cultures inoculated with cells that were sorted after stimulation with methane, Methylobacter sequences could be detected, whereas in enrichment cultures inoculated by randomly sorted cells, Methylomonas species quickly outcompeted all other types.
Similar content being viewed by others

Introduction
Lake Washington is a freshwater lake in Seattle, Washington. The upper layer of the sediment of this lake is known for high rates of methane metabolism and is an attractive model for studying environmental cycling of C1 compounds. The C1-utilizing bacterial community has been characterized so far using a number of culture-reliant (Auman et al., 2000; Miller et al., 2005; Kalyuzhnaya et al., 2005a, 2006a) and culture-independent methods, including surveys of functional genes, detection of specific RNA transcripts and stable isotope probing (Costello and Lidstrom, 1999; Kalyuzhnaya et al., 2005b; Nercessian et al., 2005). The picture emerging from these studies shows a complex multimember community that includes species known for methylotrophic capability (Methylomonas, Methylobacter, Methylocystis, Methyloversatilis, Methylotenera etc.) as well representatives of phyla not known for methylotrophy (such as Planctomycetes, Acidobacteria, Gemmatimonadetes etc.), with specific groups revealing specific nutritional preferences (Kalyuzhnaya et al., 2005c; Nercessian et al., 2005). In addition, species are present unidentified phylogenetically, being only detected at the level of functional genes (Kalyuzhnaya et al., 2005b). In an attempt to connect predicted function to phylogenetic identity, we have previously developed a method including fluorescent in situ hybridization (FISH) staining combined with flow cytometry (FC) and cell sorting (Kalyuzhnaya et al., 2006b). However, one of the main disadvantages of this method is the necessity of killing cells for FISH staining, precluding attempts of cultivation or any other downstream analyses requiring live cells. In this work, we employed redox sensor green (RSG), a novel fluorogenic redox indicator dye that recently became available from Molecular Probes, Invitrogen (Carlsbad, CA, USA) (Gray et al., 2005). This dye yields green fluorescence (488 nm excitation) when modified by bacterial reductases, many of which are parts of electron transport systems. We demonstrated here that RSG does not suppress cellular metabolism, unlike tetrazolium salts, such as 5-cyano-2,3-ditolyl tetrazolium chloride (CTC) or 2-(p-iodophenyl-3-(p-nitrophenyl)-5-phenyltetrazolium chloride (INT) (Ullrich et al., 1996) and could be used for real-time detection of cells actively respiring in situ. The advantage of combining RSG staining with FC and cell sorting is in the wealth of potential downstream applications for populations of live cells that are separated based on their specific metabolic responses.
Materials and methods
Bacterial cultures and growth conditions
Pure cultures of the following methylotrophic bacteria were used for calibration and optimization experiments: Methylobacterium extorquens AM1, Methylococcus capsulatus Bath, Methylotenera mobilis JLW8, Methylobacillus flagellatus KT and Methylomonas sp. LW13, all from the Lidstrom lab collection. M. extorquens, M. flagellatus and M. mobilis were grown in a mineral salts medium (Harder et al., 1973) termed Hypho medium, supplemented with 50 mM methanol for the two former strains and 30 mM methylamine hydrochloride for the latter. M. capsulatus and Methylomonas sp. LW13 were grown in nitrate mineral salts medium (Whittenbury et al., 1970) with a methane/air headspace ratio of 50:50. Cultures were grown at 30 °C except for M. capsulatus and M. flagellatus, which were grown at 37 °C.
Redox sensor green staining and substrate activation conditions
Redox sensor green reagent (1 μl BacLight Redox Sensor Green Vitality kit, Invitrogen) is recommended by the manufacturer for use with 106 cells per ml. We tested for optimal staining using 1 μl of RSG per 105–108 cells per ml and found that this amount of RSG is sufficient for optimal staining of up to 106 cells, independent of sample volume. For experiments using pure cultures, 2 ml samples with optical density (OD600 0.4–0.8) were mixed with 2 μl of RSG and incubated for 15–30 min at room temperature, followed by immediate analysis by microscopy or FC, as described below. Propidium iodide staining was performed using the reagent and the protocol supplied as part of the BacLight kit. For the experiments involving starvation followed by substrate activation, 50 ml samples of cultures (OD600 0.6–0.8) were harvested by centrifugation, washed with and resuspended in 50 ml of the appropriate growth medium without carbon source. Cultures were starved overnight (for approximately 12 h) at 30 °C, resulting in a significant drop (up to 100%) in the population of metabolically active cells (Figure 1). To test for substrate stimulation, 15 ml samples of the starved cultures were incubated with a test substrate at room temperature for 0.25–16 h before microscopy or FC analysis. The substrate concentrations were as follows: 0.4 mM methane (as an appropriate dilution of methane-saturated nitrate mineral salts medium), 25 mM methanol, 25 mM methylamine and 1 mM formaldehyde.

Microscopic observation and flow cytometric analysis of cells of Methylococcus capsulatus, Methylobacillus flagellatus and Methylobacterium extorquens stained with RSG for 30 min. (a) light (left) and fluorescence (right) microscopy of starved cells (top) and cells activated with methanol (bottom) after 3 h incubation. (b) Density plots (side scattering, right angle light scattering versus green fluorescence intensity) of starved cells. Top, before and after staining with redox sensor green (RSG); bottom, incubated with methanol for 1 and 3 h, respectively. Cells in gate R1 were counted and percent of total population counted is shown.
Lake Washington sediment core samples were collected as described previously (Kalyuzhnaya et al., 2005b). Sediment cores were transported to the laboratory on ice and stored at 4 °C. The samples were analyzed within the first 1–4 days after collection. Water samples were collected from above the sediment surface, sterilized by filtration immediately after transportation to the laboratory and stored at 4 °C. For FC analysis, cells were separated from the sediment particles as previously described (Kalyuzhnaya et al., 2006b). For some experiments, a modification of this protocol was employed, in which all organic extractants were omitted (see the Results section). For substrate activation, a range of concentrations was used as follows: methane (0.02–0.4 mM), methanol (0.05–50 mM), methylamine (0.05–50 mM) and formaldehyde (0.01–5 mM). The incubation time varied from 1 to 16 h at room temperature (20–22 °C). Samples (2 ml) were used for FC analysis, after staining with 2 μl of RSG for 30 min.
Viability assays
To assess cell viability, RSG-exposed samples were serially diluted as appropriate and plated onto culture-appropriate solid media supplemented with culture-appropriate growth substrates (as above), in triplicates and these were incubated at appropriate temperatures (30 or 37 °C), followed by colony counting. Samples of the same cultures not exposed to RSG served as controls.
Microscopy
Cells were observed using an epifluorescence microscope (PASCAL LSM 5, Zeiss) and a 100 × oil immersion objective.
Fluorescence-activated cytometry and cell sorting
Flow cytometric analysis was performed at the Cell Analysis Facility at the Department of Immunology (University of Washington) using a BD LSR bench-top flow cytometer (Becton Dickinson, Franklin Lakes, NY, USA). Forward angle light scattering, right angle light scattering and fluorescence of microbial cells were measured simultaneously. For each sample, 10 000 events were acquired. Data analysis was performed and graphics were acquired using the WinMDI 2.1 software package (http://facs.scripps.edu/software.html). Cell sorting was performed using a BD FACSAria high-speed four-way sorter. The instrument tubing was sterilized using, sequentially, 10% bleach, 3% hydrogen peroxide, 70% ethanol and sterile phosphate-buffered saline. Cells were sorted based on light-scattering properties (Forward angle light scattering/right angle light scattering) and green fluorescence intensity (excitation 488 nm, emission 530 nm). An unstained sediment sample (negative control) was used to define the background fluorescence. Sorting speed was maintained at 3000–6000 events s−1, with the efficiency above 80%. Cells were collected at 0.2–1 × 106 events per 12 ml tube, with a total of two tubes per experiment. One tube was used for the enrichment experiments and the second for DNA extraction.
Enrichment experiments
Randomly sorted cells, RSG-stained cells or RSG-stained cells after preincubation with methane, approximately 0.1–0.2 × 106 events in each case, were used to inoculate 10 ml of 0.1 × nitrate mineral salts medium. These were incubated in 100 ml vials, under air/methane (4:1) atmosphere, at 10 °C, agitated at 125 r.p.m. for 2 weeks, followed by transfer into a fresh medium (1 ml of culture into 24 ml of fresh medium) followed by incubation for an additional week in the same conditions, followed by the next transfer. Serial dilutions of the resulting cultures were plated onto 0.2 × nitrate mineral salts medium and plates were incubated under methane/air (50:50) at 10 °C for up to 4 weeks. Single colonies were further purified by re-streaking onto the same medium, and culture purity was monitored by microscopy. For strain identification, DNA was isolated and 16S rRNA genes were amplified and sequenced as described below.
PCR amplification, cloning and denaturing gradient gel electrophoresis profiling
DNA from the sorted cells, enrichments and pure cultures was isolated using QIAamp DNA mini kit (QIAGEN, Velno, The Netherlands). Polymerase chain reaction (PCR) amplifications of the 16S rRNA gene fragment were carried out in a total volume of 20 μl containing 0.1 μg of template DNA, 200 μM dNTPs, 10% Q-solution, 2.5 mM MgCl2, 0.2 μM universal bacterial primers Eub27f and EUB 1492r (Lane, 1991) and 0.2 U of Taq-DNA polymerase (Invitrogen). The amplification conditions were as follows: 15 min at 96 °C, 24 cycles of 40 s at 96 °C, 40 s at 56 °C and 1.5 min at 72 °C, and final extension step of 10 min at 72 °C. PCR fragments were cloned into the pCR2.1 vector (Invitrogen). Plasmid DNA was purified using Montage (Millipore, Billerica, MA, USA) or GeneJet (Fermentas, Vilnius, Lithuania) plasmid miniprep kits. Randomly selected clones (134–240) from each sorting experiment were sequenced using a forward primer (Topo TA kit, Invitrogen) and the BigDye 3.1 termination sequencing kit (Applied Biosystems, Foster City, CA, USA). Reaction analysis was performed by the Department of Biochemistry Sequencing Facility at the University of Washington (http://dnaseq.bchem.washington.edu/bdsf/).
PCR amplification of 16S rRNA gene fragments for the denaturing gradient gel electrophoresis (DGGE) profiling was carried out as described above, using primers 341F(GC) and 518R (Muyzer et al., 1993). The amplification conditions were 15 min at 96 °C, 10 cycles of 40 s at 96 °C; 40 s at 58 °C with temperature decrease by 1 °C every second cycle; 40 s at 72 °C, 14 cycles of 40 s at 96 °C, 40 s at 52 °C and 45 s at 72 °C, and a single final extension for 10 min at 72 °C. DGGE separation was performed using the DGene system (Bio-Rad, Hercules, CA, USA), in 12% polyacrylamide gels (bis-acrylamide gel stock solution, 37.5:1; Bio-Rad Laboratories, Inc.) and denaturant concentration gradient 30–70% (100% defined as 40% formamide and 7 M urea). Electrophoresis was carried out at 60 V for 16 h at 60 °C. Gels were stained with ethidium bromide (5 μg ml−1) for 15–30 min, rinsed for 10 min in water, visualized by UV and documented using a Gel Logic 100 imaging system (Kodak). The bands of interest were recovered from the DGGE gel by electrophoresis in 1% agarose gel or by elution from gel as described (Sekiguchi et al., 2001). DNA was purified using the QIAquick gel extraction kit (Qiagen) and cloned into the pCR2.1 cloning vector (Invitrogen), producing separate clone libraries for each band. Five randomly picked clones from each library were sequenced.
Results
Evaluation of redox sensor green dye performance
Redox sensor green is a novel fluorogenic redox indicator that recently became available from Invitrogen, Molecular Probes. The dye has been successfully tested on a number of bacterial species, as an indicator of vitality (Gray et al., 2005). However, its potential for detecting metabolically active cells in natural microbial populations has not yet been investigated. To evaluate this potential, we first optimized RSG staining protocols on pure culture isolates. Six methylotrophic bacteria were employed: M. capsulatus, Methylomonas sp. LW13, M. flagellatus, M. mobilis, M. universalis and M. extorquens grown as described in the Materials and methods section. Cells were first observed microscopically and then subjected to FC counting, pre- and post-incubation with RSG. Significant fractions of cells in each culture displayed a high fluorescence signal after staining with RSG (75–94%), although the intensity of fluorescence differed between the strains, cells of M. flagellatus and M. mobilis were the brightest (data not shown).
Redox sensor green staining efficiency was then evaluated as a function of physiological state of cells (active metabolism versus starvation). For these experiments, exponentially growing cells of M. capsulatus, M. flagellatus and M. extorquens were transferred into appropriate media lacking any carbon or energy source and incubated overnight to starve. Cells were then observed microscopically, and the proportion of fluorescent cells was counted by FC. For each culture, a dramatic drop occurred in the population of cells displaying fluorescent signal when stained with RSG, likely indicating a major metabolic shutdown. A growth substrate was then added to the same cells (25 mM methanol) and a steady increase in fluorescence was observed as a function of time, with the majority of cells responding after a period of approximately 3 h (Figure 1). In each case, a subpopulation of cells (10–20%) was present that did not produce a fluorescent signal even after prolonged incubation (up to 16 h) with the carbon/energy source. We tested whether these cells were dead or alive, using propidium iodide staining. Only 1–2% of cells were positively stained with propidium iodide. The majority of the cells that were negative for RSG-staining demonstrated negative staining with propidium iodide, indicating the presence of intact membranes and suggesting these cells were alive but dormant.
The dependence of the response on the substrate used for stimulation was also tested. Experiments similar to the ones described above were performed replacing methanol with methane (0.4 mM) or methylamine (25 mM). Cells of M. flagellatus and M. extorquens responded to the addition of methylamine but not methane, whereas cells of M. capsulatus responded to the addition of methane but not methylamine (Supplementary Figure 1). These data are in agreement with the known substrate range of the organisms tested: M. flagellatus and M. extorquens can oxidize both methanol and methylamine but not methane, whereas M. capsulatus can oxidize methane and methanol but not methylamine.
To test whether the cells maintained their viability after staining with RSG, 2 ml of exponentially grown cells were incubated with or without RSG for 1 h, and cell counts were determined using plating assays. Similar cell counts were obtained for cells exposed to RSG compared to nonexposed controls, demonstrating that cell viability was not significantly affected by RSG.
Application of redox sensor green for detection of actively metabolizing microbes in Lake Washington sediment and optimization of flow cytometric counting
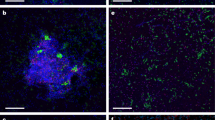
Samples of Lake Washington sediment were exposed to RSG for 15 min at room temperature and fluorescing cells were observed microscopically (Figures 2a and b), indicating that the protocol for RSG-based cell activity detection optimized on pure cultures also detected active cells in environmental samples. Our main interest was in enumerating active cells from environmental samples, determining their response to specific environmentally relevant compounds and determining their phylogenetic identity. Therefore, extracting cells from sediment particles was a necessary step. We have previously developed an efficient protocol for cell extraction involving organic extractants (sucrose and triton X-100; Kalyuzhnaya et al., 2006b) and this protocol was employed for optimization of RSG-based cell activity detection in environmental populations. The FC analysis of the extracted cells stained with RSG showed that in these conditions, a large proportion of cells (47.6±5.3%) produced a bright fluorescent signal.

Microscopic observation and flow cytometric analysis of a Lake Washington sediment sample. (a and b) light and fluorescence microscopy. (c) Flow cytometric analysis of cells (left to right) stained with redox sensor green (RSG) and cells preincubated for 1 h before staining with triton X-100, 0.2 mM methane, 0.5 mM methanol, 0.5 mM methylamine and 0.1 mM formaldehyde. R1, gate used for cell counting and sorting.
Optimization of cell extraction methods for detection of substrate-activated respiration
It has been previously demonstrated by CTC staining that exposure to organic compounds increased the number of respiring cells in a population, as a result of substrate stimulation (Gruden et al., 2003). RSG was tested for utility in detecting specific response of microbial populations to C1 substrate stimuli. For these experiments, the cell extraction protocol used for the experiments described above was modified by omitting all organic extractants from the elution buffer, to eliminate any potential effect of the extractants on cell respiration. This modification resulted in a dramatic decrease in cell extraction efficiency, of approximately two orders of magnitude. Cells extracted this way were subjected to RSG staining and FC as described above. In this case, a much smaller fraction of cells (13.8±0.8%) produced high-fluorescence signal (Figure 2c). To test whether the drop in the population of active cells was due to the difference in the organic content between the original and the modified elution buffer, cells extracted from the sediment particles using the modified buffer were incubated with either sucrose or triton X-100 for 10–60 min at room temperature. Cells from the same extraction batch incubated without any additions served as a control. These were then stained with RSG and subjected to FC. We found that incubation with sucrose had no significant effect on the count of fluorescent cells compared with the control, increasing by 2.1±0.2% (data not shown). However, cells incubated with triton X-100 even for a short period of time (10–15 min) displayed much higher counts of fluorescent events (50±3%; Figure 2c). This number was in good agreement with the number obtained for cells eluted with triton X100-supplemented buffer (see above). Since triton X-100 is not a likely carbon/energy substrate (Zeng et al., 2007), the sharp increase in the number of RSG-stained cells is most probably not due to metabolic activation but due to the surfactant properties of triton X-100, affecting the permeability of the cell membrane (Helenius and Simons, 1975; Nemec and Jernejc, 2002) and facilitating cell penetration by RSG. These results indicated that to use this approach to obtain cells for downstream analysis, including cultivation, a modification of the extraction buffer omitting triton X-100 was necessary to avoid the risk of cell membrane disruption.
Use of redox sensor green for detection of substrate-activated respiration
Four different C1 substrates, methane, methanol, methylamine and formaldehyde, were tested as potential stimuli of metabolic activity in the sediment community, with a range of concentrations tested for each stimulus (Supplementary Table 1). Two different stimulation protocols were tested, adding each stimulus either before or after cell extraction from sediment particles. In either case, a statistically significant increase in the number of fluorescing cells was observed. The response was more pronounced in the lower range of substrate concentrations, with a higher number of cells responding and faster maximum response observed. These results suggest an inhibitory effect for higher concentrations of the substrates used. However, the lowest concentrations tested (in the 0.01–0.05 mM range) did not result in stimulatory effect, suggesting that a threshold exists for noticeable effect of a stimulus (Supplementary Table 1). To summarize these data, 13.8±0.8% of the total cell population produced a fluorescent signal without substrate stimulation and maximal stimulation was achieved using 0.2 mM methane (23.9±4.2%), 0.5 mM methanol (17.0±0.2%), 0.5 mM methylamine (19.7±0.2%) and 0.1 m formaldehyde (20.1±0.5%) (Figure 2c and Supplementary Table 1), with the initial response detected approximately 30 min after the addition of the substrate and the maximum response reached in approximately 3 h. The smaller numbers of cells responding to methanol and formaldehyde compared to that of cells responding to methane may be due to inhibitory effects of these substrates on some methane-oxidizing species.
Substrate-specific enrichment and phylogenetic profiling of populations responding to C1 substrates
It seemed likely that cells specifically responding to the addition of C1 substrate stimuli are involved in metabolism of C1 substrates. Therefore, RSG was tested for utility in targeted analysis of such cells. Cells extracted from the sediment were RSG-labeled and incubated with no substrate, methane, methanol, methylamine and formaldehyde for 2 h at room temperature, respectively. These samples plus a sample not incubated with RSG were then subjected to FC and cell sorting as previously described for FISH-labeled cells (Kalyuzhnaya et al., 2006b). Approximately 0.1–1 × 106 fluorescent events or randomly sorted cells (control sample) were collected from each experiment, and the resulting material was used for DNA extraction, followed by PCR amplification of 16S rRNA gene fragments. These were subjected to DGGE analysis and the resulting DGGE profiles were compared (Figure 3). From these comparisons, several phylotypes were identified that were only present in substrate-activated cell populations and were not detectable at the total community level. Conversely, several phylotypes were found at the total community level that were not detectable in substrate-activated cell fractions. In addition, the DGGE profiles resulting from different C1-compound activation experiments, each revealed specific patterns. The DGGE pattern of cells activated with methylamine was most distinct, potentially indicating that not only methylotrophs but also non-methylotrophic species that are nitrogen-limited may be responding. However, as this work is a proof-of-concept study, the nature of the stimulus is less important than the fact that bacterial response to the stimulus can be observed and monitored. Major DGGE bands for each pattern were extracted from gels, and the resulting DNA fragments sequenced. The three most prominent bands in the methane-responding fraction represented Methylomonas sp. LW21, an organism originally isolated from the site, an unclassified deltaproteobacterim and an unclassified alphaproteobacterium. The most prominent band in the methanol-responding fraction represented an Acidovorax phylotype. Acidovorax are metabolically versatile members of Comamonadaceae, a family containing bona fide methylotrophs (Anesti et al., 2005; Nakatsu et al., 2006) and the specific presence of this sequence may point toward the existence of yet uncultivated methylotrophic Acidovorax. The three most prominent bands in the methylamine-responding fraction were represented by sequences most closely related to, respectively, Xanthomonaceae, Comamonadaceae and Afipia, the latter also containing methylotrophic representatives (Moosvi et al., 2005). The major bands in the formaldehyde-responding fraction represented an unclassified betaproteobacterium, an unclassified deltaproteobacterium and a Sphingobacteriales bacterium (Supplementary Figure 2).

Denaturing gradient gel electrophoresis (DGGE) analysis of 16S rRNA gene fragments amplified from randomly sorted cells (a) and cells sorted after staining with redox sensor green (RSG) (b–e). (b) No substrate stimuli, (c) stimulated by methane, (d) stimulated by methanol and (e) stimulated by methylamine. The sequences of DGGE bands are as follows: 1, Methylomonas sp.; 2, unclassified deltaproteobacterium; 3, unclassified alphaproteobacterium; 4, Acidovorax sp.; 5, Xanthomonadaceae; 6, Comamonadaceae; 7, Afipia sp. Additional bands specific to each sample are indicated by slim arrows. Triplicate fingerprints are shown.
The composition of the community responding to methane was investigated in more detail. 16S rRNA gene fragments from the sorted cells were PCR-amplified, followed by library construction, sequencing of 134 randomly selected clones and sequence analysis. These data were compared to the data generated in a similar way for the non-enriched sediment sample (240 clones) and to the fraction of cells metabolically active before the addition of substrate stimuli (180 Clones). Representation of major bacterial phyla was assessed and compared among the three libraries, as follows. The non-enriched sample was dominated by Betaproteobacteria, Deltaproteobacteria, Acidobacteria and Bacteroidetes (23, 14, 13 and 10% of total sequences; Figure 4 and Supplementary Table 2). Within the Proteobacteria, the most represented groups were Rhodocyclaceae, Burkholderiaceae, and Syntrophonabacterales. Whereas the actively respiring fraction (positive for RSG staining) was also dominated by Betaproteobactera (29%), certain shifts in representation of other groups were observed as follows. Gemmatimonadetes, Verrucomicrobiales and Firmicutes were more represented compared to the non-enriched library (8% versus 3%; 8% versus 5% and 3% versus 1%, respectively), whereas Acidobacteria were less represented 5% versus 13%). Shifts were also observed within the proteobacterial population, the most represented families being Commamonadaceaea, Xanthobacteriaceae, Rhodobacteraceae, Methylophilaceae, Nitrosomonadaceae and Xanthomonadaceae and a diverse group of bacteria related to unclassified Burkholderiales. The families Alcaligenaceae, Rhodocyclaceae and Desulfobacteriaceae were underrepresented within the RSG-positive population (Supplementary Table 1). The composition of the community responding to methane was distinct from the RSG-exposed control. The proportion of Methylococcaceae sequences increased fourfold, to 12% of the total population, and the proportion of sequences related to uncultured and unclassified Deltaproteobacteria increased fivefold, to 16% of total population. The enriched deltaproteobacterial sequences were only distantly related to representatives of Anaeromyxobacter dehalogenans (<88% sequence similarity), Desulfuromonadales and Desulfobacteraceae. Other groups abundance of which increased, to a lesser degree, were uncultured Verrucomicrobiales, uncultured Acidobacteria and bacteria represented by unaffiliated sequences. Of all the sequences uncovered by the analysis of the methane-activated cells, only the Methylococcaceae sequences were closely related to the sequences of cultivated organisms, Methylobacter tundrapulidum (96–99% sequence similarity) and Methylomonas methanica (96–97% similarity). Methylomonas species are readily cultivable from the site (Auman et al., 2000). However, so far no Methylobacter has been cultivated, despite significant efforts toward expanding the range of cultivated methylotrophs from Lake Washington (Auman et al., 2000 and this study, see below).

Taxonomic distribution of bacterial phyla in 16S rRNA gene libraries prepared from randomly sorted cells (1), sorted redox sensor green (RSG)-stained cells (2) and sorted methane-activated RSG-stained cells (3). Arrows indicate phyla whose presence significantly increased after stimulation with methane.
Culture enrichment experiments
The methane-stimulated subpopulations were used in enrichments, to determine whether the presorting enhanced the ability to enrich for species reluctant to cultivation, such as the Lake Washington Methylobacter that has been detected by culture-independent experiments (in this study and Nercessian et al., 2005) but never cultivated. Three types of enrichment cultivations were set up. One flask was inoculated with approximately 0.1 × 106 randomly sorted cells representing the non-enriched total population, the second was inoculated by the same number of cells that were metabolically active before methane stimulation and the third was inoculated by the same number of methane-stimulated cells. The cultures were incubated at 10 °C in a methane/air atmosphere for 2 weeks with shaking, followed by two transfers, as described in the Materials and methods section. The resulting cultures were used for DNA extraction. 16S rRNA genes were PCR-amplified, followed by DGGE analysis and the DGGE profiles were compared between the three types of enrichments (Figure 5). Based on these analyses, the enrichment culture inoculated by randomly sorted cells was dominated by Methylomonas spp. and Methylosarcina lacus, the enrichment culture inoculated by active (RSG-positive) cells was dominated by Methylocystis sp., with a minor presence of Methylomonas and M. lacus, and the enrichment culture inoculated by methane-activated cells revealed the presence of a variety of phylotypes, including Methylomonas, Methylocystis, Methylosinus, M. lacus and Methylobacter. Representatives of the former five genera have been previously isolated from Lake Washington sediment, by routine enrichments (Auman et al., 2000; Kalyuzhnaya et al., 2006a). In this study, we obtained pure cultures of all the Methylomonas strains represented by prominent DGGE bands, as well as the Methylocystis strain represented by a single DGGE band (noted by asterisks in Figure 5). However, so far no pure cultures of Methylobacter sp. detected in this study and in previously described culture-independent experiments (Nercessian et al., 2005) were isolated from the site.

Denaturing gradient gel electrophoresis (DGGE) analysis of 16S rRNA gene fragments amplified from randomly sorted cells (1), sorted redox sensor green (RSG)-stained cells (2), sorted methane-activated RSG-stained cells (3) and from methane enrichment cultures inoculated with randomly sorted cells (4), sorted RSG-stained cells (5) and sorted methane-activated RSG-stained cells (6).
Discussion
Microbial communities inhabiting most natural environments are very complex, supporting a variety of biogeochemical processes specific to each environmental niche. Because most environmental microbes are uncultivated and unknown (Rappe and Giovanoni, 2003; Schloss and Handelsman, 2005; Green and Keller, 2006), methods for culture-independent detection and analysis of environmental microbes and for understanding their specific roles are very important. This is especially true for methods directly linking specific metabolic activities to specific phylotypes in a given environment, such as DNA and RNA stable isotope probing (Radajewski et al., 2000; Manefield et al., 2002), selective nucleic acid polymerase-biotinylation and capture (Borneman 1999; Van Mooy et al., 2004), isotope ratio mass spectrometry (Eek et al., 2007) or secondary ion spectrometry (Orphan et al., 2001). Here, we presented a novel culture-independent method for detecting, observing and manipulating environmental microbes, based on their respiratory activity as detected by RSG, a novel fluorogenic dye from Invitrogen. This is the first work, to our knowledge, in which this dye was used to detect and enumerate microbes actively metabolizing in situ, in real time. It was also possible to manipulate these microbes to observe specific response to the addition of test substrates relevant to metabolic activities taking place in an environment of interest. In this case, microbial response to the addition of C1 substrates, methane, methanol, methylamine and formaldehyde in Lake Washington sediment community was followed, and specific shifts in community structure were demonstrated using DGGE.
As a proof of concept, the community responding to methane was investigated, determining that it was made up of not only of bona fide gamma- and alphaproteobacterial methanotrophs previously studied in cultures, but also of representatives of other phylogenetic groups (Deltaproteobacteria, Verrucomicrobiales and Acidobacteria), with no cultivated representatives. These results suggest that these organisms may be involved in metabolism of methane in the site, either directly or indirectly. Indeed, methanotrophic Verrucomicrobiales have been described recently from acidic environments (Dunfield et al. 2007; Pol et al., 2007; Islam et al., 2008) and our data suggest the existence of methanotrophic Verrucomicrobiales in freshwater lakes.
We also determined that RSG staining-based cell separation may be useful in facilitating cultivation of at least some of the microbes with relevant activities, by increasing their relative abundance. This is especially interesting with regard to the Methylobacter species detected in Lake Washington by culture-independent approaches (Nercessian et al., 2005) that have so far resisted cultivation, which were identified in enrichment cultures inoculated with cells sorted for RSG signal after methane stimulation. It is possible that they require certain nutrients missing from the medium typically used for methanotroph enrichment (Whittenbury et al., 1970). However, the experiments described here suggest it is more likely that Methylobacter species are outcompeted by other methanotrophs, such as Methylomonas. Sorting by methane-induced respiratory activity apparently provides numeric advantage to Methylobacter, resulting in enrichment for their sequences. Attempts are in progress to isolate pure cultures of Methylobacter species represented by the sequences uncovered in this study.
The method described here should be applicable to a wide variety of microbes and substrates, as it relies only on respiratory activity. The advantages of RSG compared to other stains utilized to detect microbial activity (for example, CTC) are the rapid response to redox activity within the cell, stable fluorescence signal, quick cell penetration and the lack of detrimental effect on core cell functions. Any substrate that can be transported into the cell can be used as a stimulus if substrate-specific response is desired. Besides PCR amplification of genes of interest and cultivation attempts, other potential downstream applications for cell populations sorted based on activity stain include metagenomic sequencing and metaproteomic analysis. Our results demonstrate that RSG coupled with microscopy, FC and cell sorting is a valuable tool in the effort to link function with phylotype in environmental microbial communities. Although cell extraction is necessary for cell counting and separation from sediment or soil samples, cells from environments, such as water columns or enrichment cultures, can be counted directly, making the method even more straightforward.
Although the advantages of the method described here are clear, a warning should be given on its potential limitations, the most likely ones including RSG transport problems that may be encountered by some species, requirement for relatively high stimulus concentrations in cases when observation of a substrate-specific response is desired and potential inhibitory effects of some substrate stimuli. Thus, the RSG-based approach described here should be most informative when combined with other culture-independent methods, such as DNA- or RNA stable isotope probing.
References
Anesti V, McDonald IR, Ramaswamy M, Wade WG, Kelly DP, Wood AP . (2005). Isolation and molecular detection of methylotrophic bacteria occurring in the human mouth. Environ Microbiol 7: 1227–1238.
Auman AJ, Stolyar S, Costello AM, Lidstrom ME . (2000). Molecular characterization of methanotrophic isolates from freshwater lake sediment. Appl Environ Microbiol 66: 5259–5266.
Borneman J . (1999). Culture-independent identification of microorganisms that respond to specified stimuli. Appl Environ Microbiol 65: 3398–3400.
Costello AM, Lidstrom ME . (1999). Molecular characterization of functional and phylogenetic genes from natural populations of methanotrophs in lake sediments. Appl Environ Microbiol 65: 5066–5074.
Dunfield PF, Yuryev A, Senin P, Smirnova AV, Stott MB, Hou S et al. (2007). Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450: 879–882.
Eek KM, Sessions AL, Lies PD . (2007). Carbon-isotopic analysis of microbial cells sorted by flow cytometry. Geobiol 5: 89–95.
Gray D, Yue RS, Chueng CY, Godfrey W . (2005). Bacterial vitality detected by a novel fluorogenic redox dye using flow cytometry. In: Abstracts of the American Society of Microbiology Meeting. American Society for Microbiology, Washington, DC, USA, http://probes.invitrogen.com/servlets/posters/.
Green BD, Keller M . (2006). Capturing the uncultivated majority. Curr Opin Biotechnol 17: 236–240.
Gruden CL, Khijniak A, Ariaens P . (2003). Activity assessment of microorganisms eluted from sediments using 5-cyano-2,3-ditolyl tetrazolium chloride: a quantitative comparison of flow cytometry to epifluorescent microscopy. J Microbiol Methods 55: 865–874.
Harder W, Attwood M, Quayele JR . (1973). Methanol assimilation by Hyphomicrobium spp. J Gen Microbiol 78: 155–163.
Helenius A, Simons K . (1975). Solubilization of membranes by detergents. Biochim Biophys Acta 415: 29–79.
Islam T, Jensen S, Reigstad LJ, Larsen O, Birkeland NK . (2008). Methane oxidation at 55 degrees C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. Proc Natl Acad Sci USA 105: 300–304.
Kalyuzhnaya MG, Bowerman S, Lara JC, Lidstrom ME, Chistoserdova L . (2006a). Methylotenera gen. nov., a new genus in the family Methylophilaceae, and description of Methylotenera mobila, an obligate methylamine utilizing bacterium. Int J Syst Evol Microbiol 56: 2819–2823.
Kalyuzhnaya MG, Bowerman S, Nercessian O, Lidstrom ME, Chistoserdova L . (2005b). Highly divergent genes for methanopterin-linked C1 transfer reactions in Lake Washington, assessed via metagenomic analysis and mRNA detection. Appl Envir Microbiol 71: 8846–8854.
Kalyuzhnaya MG, Nercessian O, Lidstrom ME, Chistoserdova L . (2005c). Development and application of polymerase chain reaction primers based on fhcD for environmental detection of methanopterin-linked C1 metabolism in bacteria. Environ Microbiol 7: 1269–1274.
Kalyuzhnaya MG, Stolyar SM, Auman AJ, Lara JC, Lidstrom ME, Chistoserdova L . (2005a). Methylosarcina lacus sp. nov., a methanotroph from Lake Washington, Seattle, USA, and emended description of the genus Methylosarcina. Int J Syst Evol Microbiol 55: 2345–2350.
Kalyuzhnaya MG, Zabinsky R, Bowerman S, Baker DR, Lidstrom ME, Chistoserdova L . (2006b). Fluorescence in situ hybridization-flow cytometry-cell sorting-based method for separation and enrichment of type I and type II methanotroph populations. Appl Environ Microbiol 72: 4293–4301.
Lane DJ . (1991). 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds.). Nucleic Acid Techniques in Bacterial Systematics. Wiley: New York, pp 115–175.
Manefield M, Whiteley AS, Griffiths RI, Bailey MJ . (2002). RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl Environ Microbiol 68: 5367–5373.
Miller JA, Kalyuzhnaya MG, Noyes E, Lara JC, Lidstrom ME, Chistoserdova L . (2005). Labrys methylaminiphilus, sp. nov., a new facultatively methylotrophic bacterium from a freshwater lake sediment. Int J Syst Evol Microbiol 55: 1247–1253.
Moosvi SA, Pacheco CC, McDonald IR, De Marco P, Pearce DA, Kelly DP et al. (2005). Isolation and properties of methanesulfonate-degrading Afipia felis from Antarctica and comparison with other strains of A. Environ Microbiol 7: 22–33.
Muyzer G, de Waal EC, Uitterlinden A . (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700.
Nakatsu CH, Hristova K, Hanada S, Meng XY, Hanson JR, Scow KM et al. (2006). Methylibium petroleiphilum gen. nov., sp. nov., a novel methyl tert-butyl ether-degrading methylotroph of the Betaproteobacteria. Int J Syst Evol Microbiol 56: 983–989.
Nemec T, Jernejc K . (2002). Influence of Tween 80 on lipid metabolism of an Aspergillus niger strain. Appl Biochem Biotech 101: 229–238.
Nercessian O, Noyes E, Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L . (2005). Bacterial populations active in metabolism of C1 compounds in the sediment of Lake Washington, a freshwater lake. Appl Envir Microbiol 71: 6885–6899.
Orphan VJ, House CH, Hinrichs K-U, McKeegan KD, DeLong EF . (2001). Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science 293: 484–487.
Pol A, Heijmans K, Harhangi HR, Tedesco D, Jetten MSM, Op den Camp HJM . (2007). Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature 450: 874–878.
Radajewski S, Ineson P, Parekh NR, Murrell JC . (2000). Stable-isotope probing as a tool in microbial ecology. Nature 403: 646–649.
Rappe MS, Giovanoni SJ . (2003). The uncultured microbial majority. Annu Rev Microbiol 57: 369–394.
Schloss PD, Handelsman J . (2005). Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biology 6: 229.
Sekiguchi H, Tomioka N, Nakahara T, Uchiyama H . (2001). A single band does not always represent single bacterial strains in denaturing gradient gel electrophoresis analysis. Biotechnol Lett 23: 1205–1208.
Ullrich S, Karrasch B, Hoppe H, Jeskulke K, Mehrens M . (1996). Toxic effects on bacterial metabolism of the Redox Dye 5-cyano-2,3-ditolyl tetrazolium chloride. Appl Environ Microbiol 62: 4587–4593.
Van Mooy BAS, Devol AH, Keil RG . (2004). Quantifying 3H-thymidine incorporation rates by a phylogenetically defined group of marine planktonic bacteria (Bacteriodetes phylum). Environ Microbiol 6: 1061–1069.
Whittenbury R, Phillips KC, Wilkinson JF . (1970). Enrichment, isolation and some properties of methane-utilizing bacteria. J Gen Microbiol 61: 205–218.
Zeng G, Fu H, Zhong H, Yuan X, Fu M, Wang W et al. (2007). Co-degradation with glucose of four surfactants, CTAB, Triton X-100, SDS and Rhamnolipid, in liquid culture media and compost matrix. Biodegrad 18: 303–310.
Acknowledgements
We are grateful to SR Levine and E Latypova (University of Washington) for assistance in sequencing and to V Karpiak (Molecular Probes) for helpful suggestions on RSG use. We acknowledge the UW Immunology/Cell Analysis Facility team for assistance in cell-sorting experiments. This work was funded by the National Science Foundation as part of the Microbial Observatories Program (MCB-0131957).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary information
Rights and permissions
About this article
Cite this article
Kalyuzhnaya, M., Lidstrom, M. & Chistoserdova, L. Real-time detection of actively metabolizing microbes by redox sensing as applied to methylotroph populations in Lake Washington. ISME J 2, 696–706 (2008). https://doi.org/10.1038/ismej.2008.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.32
Keywords
This article is cited by
-
Microbe combined with Fe2+-heat activated persulfate to decompose phenanthrene in red soil: comparison of acid-resistant degrading microflora and indigenous bacteria
Environmental Science and Pollution Research (2023)
-
Decoupling of respiration rates and abundance in marine prokaryoplankton
Nature (2022)
-
Flow cytometric approach to evaluate the impact of hydro-technical concrete compounds’ release to the freshwater microbiome
Environmental Monitoring and Assessment (2021)
-
Reciprocal growth control by competitive binding of nucleotide second messengers to a metabolic switch in Caulobacter crescentus
Nature Microbiology (2020)
-
Next-generation physiology approaches to study microbiome function at single cell level
Nature Reviews Microbiology (2020)
