
Abstract
Expression of the functional gene tfdA involved in degradation of phenoxyacetic acids such as 2,4-dichlorophenoxyacetic acid (2,4-D) and 4-chloro-2-methylphenoxyacetic acid (MCPA) was investigated during degradation scenarios in natural unseeded soil samples. The results illustrate how messenger RNA (mRNA)-based analysis is well suited to quantitatively study the activity of specific microbial populations in soil using phenoxyacetic acid biodegradation as a model system. Via quantitative real-time PCR, a clear response to the presence of phenoxy acids was shown during degradation in soil amended with 20 mg 2,4-D or MCPA per kg soil. Further, we found a relatively high degree of correlation between expression of the functional gene and the rates of mineralization. Melting curve analyses of real-time PCR products, supported by tfdA-denaturing gradient gel electrophoresis analysis showed that, although only class I tfdA genes were apparent in the indigenous microbial population, class III tfdA genes became predominant during incubation, and were the only genes expressed during degradation of MCPA in soil. In contrast, both classes were expressed during degradation of the structurally similar compound 2,4-D. The ability to quantify microbial transcripts directly in environmental samples will have a profound impact on our understanding of microbial processes in the environment in future studies.
Similar content being viewed by others


Introduction
The key to studying gene expression quantitatively in environmental samples lies in the ability to effectively and specifically detect messenger RNA (mRNA) corresponding to the activity of interest in a given sample. To date, however, studies describing the detection and quantification of mRNA in indigenous populations in complex environmental matrices are extremely rare. Two major technical problems impede this ability. First, compared to in vitro studies, ecologically important microbial activities in situ may be low in magnitude, correspondingly reflected in relatively low abundance of the corresponding mRNA transcript. Second, effective mRNA recovery and purification have been difficult due to ubiquitous RNase activities and the binding of mRNA to soil constituents (Deutscher, 2006). Nevertheless, a few studies have successfully detected gene expression within indigenous bacterial populations in natural soil, but not quantitatively (Nogales et al., 2002; Leininger et al., 2006). Where gene expression in soil has been quantified, it has typically been in samples seeded with a specific bacterial inoculum containing the functional gene of interest (Han and Semrau, 2004; Jacobsen and Holben, 2007; Nicolaisen et al., 2008). Recent improvements in mRNA extraction protocols (Nicolaisen et al., 2008) and real-time PCR detection assays for the tfdA gene (Bælum et al., 2006) have made identification and quantification of actively expressed specific functional genes in indigenous microbes more tractable.
The herbicidal compounds included in the group of phenoxyacetic acids have been among the best studied environmentally hazardous compounds in recent decades. In particular, the bacteria and genes involved in degradation of 2,4-Dichlorophenoxyacetic acid (2,4-D) have been intensively studied, while those involved in metabolizing other phenoxyacetic acids, for example, 4-chloro-2-methylphenoxyacetic acid (MCPA) are less well known. Numerous phylogenetically diverse bacteria using 2,4-D as the sole source of carbon have been isolated (Pemberton et al., 1979; Tonso et al., 1994; Kamagata et al., 1998; Itoh et al., 2000) and functional genes involved in the entire degradation pathway have been characterized (Don et al., 1985; Fukimori and Hausinger 1993). The first step in the degradation pathway is catalyzed by an α-ketoglutarate-dependent dioxygenase encoded by the tfdA gene (Figure 1). Prior work has shown that diversity among the functional tfdA genes encoding this dioxygenase enzyme is high (Vallaeys et al., 1996; Kamagata et al., 1998). McGowan et al. (1998) suggested classifying tfdA genes into three classes (I–III) with sequences between classes showing 75–80% homology and Kamagata et al. (1998) and Itoh et al. (2000) isolated 2,4-D-degrading strains possessing the tfdA-like genes tfdAα and cadA. Although bacteria and functional genes involved in MCPA degradation have been less intensively studied, several organisms isolated using 2,4-D as the sole source of carbon have been shown to degrade MCPA as well (Smejkal et al., 2001).

The catabolic activity of the TfdA enzyme in the degradation of 2,4-dichlorophenoxyacetic acid (2,4-D) and 4-chloro-2-methylphenoxyacetic acid (MCPA). α-KG, α-ketoglutarate; X, Cl- in 2,4-D and a methyl-group in MCPA; DCP, 2,4-dichlorophenol; MCP, 2-methyl-4-chlorophenol.
A number of molecular methods for microbial community analyses in complex soil samples have enabled studies of microbial populations in the context of their environment. These methods have primarily been used to describe microbial community composition and the genetic potential for specific functions based on the presence of target DNA sequences. A major caveat is that DNA analyses target not only active microorganisms, but also inactive microorganisms (Lindahl, 1993). For example, based on functional gene DNA sequences, Bælum et al. (2006) showed that in a soil dominated by bacteria carrying class I tfdA genes, only bacteria harboring class III tfdA genes were able to proliferate during degradation of MCPA. However, in this DNA-based study, it was not possible to tell whether the class I tfdA gene was actually expressed in the standing population of bacteria harboring this gene in the presence of MCPA, or if these bacteria were inactive for MCPA degradation. Hence, methodologies to detect and quantify actual gene expression related to specific microbial functions in soil environments are sorely needed.
Herein we report an initial example of quantifying specific mRNA in natural soil samples by indigenous bacterial populations and at the same time analyze the diversity of the tfdA genes and gene transcripts to indicate which classes of tfdA genes are present and which types are expressed during 2,4-D and MCPA biodegradation. To accomplish this, small microcosms of agricultural soil were treated with 14C-2,4-D or 14C-MCPA at environmentally relevant concentrations, after which 14CO2 evolution was followed over time. The soils were re-amended after complete initial mineralization so that two cycles of biodegradation were monitored. Soil samples, taken at appropriate time points, were subjected to co-extraction of total DNA and total RNA pools from the very same sample. By quantifying copy numbers of both tfdA DNA and tfdA mRNA, we calculated gene expression levels per gene copy number (as the ratio of mRNA:DNA). Furthermore, melting curve analysis of real-time PCR products and denaturing gradient gel electrophoresis (DGGE) using the same tfdA primers were used to investigate the diversity of the expressed functional genes.
Materials and methods
Bacterial strains and reference genes
Cupriavidus necator JMP134(pJP4) (Pemberton et al., 1979), Burkholderia sp. RASC (Fulthorpe et al., 1995) and an unclassified bacterial strain (Tonso et al., 1994) were used for positive controls in tfdA-DGGE and in melting curve analyses. C. necator JMP134(pJP4) was used for standard curve preparation in the real-time PCR assay. All of the bacterial strains were propagated in mineral medium (MMO) (Stanier et al., 1966) supplemented with 500 mg l−1 of 2,4-D. DNA sequence analysis confirmed that these strains contained the tfdA class I, II and III genes, respectively.
Soil sample
The soil was an agricultural topsoil from Sjællands Odde, Zeeland, Denmark and was classified as a Typic Argiudoll (USDA Soil Taxonomy) containing 19% clay, 18% silt, 62% sand and 1.2% carbon with a pH of 7.2. Approximately 100 kg soil in total was obtained using a manual composite sampling technique in which subsamples were taken from scattered locations within a 10 m2 area, mixed thoroughly and stored frozen at −20 °C in aliquots of ∼2 kg. Prior to experiments, aliquots were thawed and acclimatized in the dark at 10 °C for 10 days as described by Mortensen and Jacobsen, 2004.
Microcosm setup and phenoxy acid mineralization analysis
Triplicate microcosms were set up in 100 ml glass flasks with airtight lids. In total, 22.1 g of soil wet weight−corresponding to 20 g soil dry weight was placed inside each flask and 2,4-D or MCPA was added as a solution in Milli-Q (Milliepore, Birkerød, Denmark) water to a final concentration of 20 mg kg−1 (corresponding to 8.8 mg C kg−1 soil for 2,4-D and 10.8 mg C kg−1 soil for MCPA) and a soil moisture content of ∼70% of the water-holding capacity (WHC). Radioactive substrates were added as 50 000 d.p.m. U ring 14C-labeled 2,4-D or MCPA (radiochemical purity of >95%; Izotop, Budapest, Hungary) mixed with unlabeled 2,4-D or MCPA (chemical purity of 97.5%; Dr Ehrenstorfer GmbH, Augsburg, Germany). Mineralization was measured as 14CO2 trapped in 0.5 M NaOH as previously described (Mortensen and Jacobsen, 2004). After mineralization of the first amendment had ceased, an additional amendment of 20 mg kg−1 2,4-D or MCPA with the same 14C/12C ratio was added to the microcosms, raising the water content to 90% of WHC. Mass balances for this type of experiment have previously been determined and 14C recoveries of 93±1.5% were obtained (Andersen et al., 2001). During the periods of mineralization, subsamples of soil were collected at appropriate intervals, immediately frozen in liquid nitrogen and subsequently stored at −80 °C until nucleic acid extraction. Triplicate sampling was performed as one composite sample (that is, 5–10 small samples were taken from scattered locations in one flask and pooled) of 500 mg soil from each of the triplicate microcosms at each time point.
DNA and RNA extraction and cDNA synthesis
DNA and RNA were co-extracted as described by Nicolaisen et al. (2008) with the following modification: 1 μl of glycogen (Roché, Basel, Switzerland) was added to PEG 6000 to aid in nucleic acid precipitation. Samples were kept on ice between all steps of the extraction. Consistency of the general nucleic acid extraction efficiency was checked running 5 μl aliquots on standard 1.5% agarose gel stained with ethidium bromide. After the extraction procedure, an 8 μl aliquot of each sample was immediately used for RNase-free DNase I treatment (Promega, Madison, WI, USA) according to the manufacturer's protocol. After the DNase I treatment, an aliquot of 4 μl was used as template in a reverse transcription (RT) reaction using the Omniscript reverse transcription kit from Qiagen (Crawley, UK) and random hexamer primers from Promega. RT was performed as a 90 min reaction at 37 °C terminated by 5 min incubation at 93 °C.
Quantitative real-time PCR
Standards with known quantities of the bacterium C. necator JMP134 harboring the class I tfdA gene were made by addition of 100 μl of washed 10-fold dilutions of C. necator JMP134 cells in 0.015 M phosphate buffer (pH 7.4) to five microcentrifuge tubes containing 500 mg soil to produce final cell concentrations of 3 × 107, 3 × 106, 3 × 105, 3 × 104, 3 × 103 colony-forming units per gram soil. DNA was extracted from these seeded soils as described above. Real-time PCR quantification as well as melting curve analysis were performed as previously described (Bælum et al., 2006) except that primers forward: 5′-GAG CAC TAC GC(AG) CTG AA(CT) TCC CG-3′ and reverse: 5′-GTC GCG TGC AGA AG-3′ were used and QuantiTect SYBR Green PCR kit (Qiagen) was replaced by DyNAmo SYBR Green qPCR kit (FinnZymes, Helsinki, Finland). All DNA extracts were diluted 10-fold prior to use in PCR reactions to reduce possible humic acid interference, while the cDNA samples resulting from the DNase digestion and RT steps were only diluted 5-fold. qPCR reactions were performed in triplicate on all DNA samples, cDNA samples and DNase-treated RNA samples. The qPCR reactions using DNase-treated RNA samples were performed as control reactions for possible DNA contamination that would interfere with cDNA-based quantification of gene expression levels. Although confirmatory melting curve analysis was performed on all real-time PCR products, only a select few are presented below for simplicity and clarity. These were selected to represent points before, at the onset of, and late in the degradation phase.
Denaturing gradient gel electrophoresis
For DGGE analysis, template from the same sampling points described above for melting curve analysis was used for PCR amplification. Briefly, PCR was performed in 50 μl reactions containing dH2O, buffer, 1.5 mM MgCl2, 200 μM deoxyribonucleotide triphosphate mix, iTaq DNA polymerase (Bio-Rad, Hercules, CA, USA), 0.4 μM HdA primers as above with a GC-clamp as described in Bælum et al. (2006), 51 μg bovine serum albumin (New England Biolabs, MA, USA) and 1 μl DNA or cDNA template. Reactions were performed in an iCycler Thermocycler (Bio-Rad) and conditions were as follows: 95 °C for 15 min followed by 40 cycles of 1 min at 95 °C, 1 min at 64 °C and 6 min at 72 °C.
DGGE analyses were performed by electrophoresis on 8% polyacrylamide gels containing a urea-formamide gradient of 55–70% (where 100% denaturant contained 7 M urea and 40% formamide). Electrophoresis was carried out at 130 V for 5 h in 1 × Tris-acetate-EDTA buffer using a D-code apparatus (Bio-Rad). After electrophoresis, the gels were stained with SYBR gold (Molecular probes, Eugene, OR, USA) for 45 min, followed by a brief rinse with Milli-Q water and then visualized under UV light and finally photographed. DNA bands of interest were excised from the gels, re-amplified using non-clamped primers and sequenced on an ABI3130x sequencer (Applied Biosystems, Foster City, CA, USA). The resultant sequences were manually aligned with the three reference tfdA sequences. Sequences are deposited in GenBank under deposition numbers EF648603 through EF648609 for selected bands (Figure 5, bands 1–7).
Data analysis
Cumulative mineralization data were corrected for background radioactivity and for the loss of radioactivity associated with removal of 500 mg soil when samples for nucleic acid extraction were taken. Nonlinear regression analysis was performed on mineralization data using the curve-fitting program in Sigmaplot (version 10.0, SPSS) as previously described (Mortensen and Jacobsen, 2004). Briefly, the data were fitted to a model describing first order kinetics and to the two 3/2-order models developed by Brunner and Focht (1984), where one model assumes linear or zero growth and the other assumes exponential growth of microorganisms during the mineralization. Furthermore, quantities of tfdA mRNA and tfdA gene copy number were related to the mineralization rate by the differentiated form of the 3/2-order model best describing the data.
Results
Mineralization of phenoxy acids
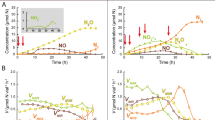
Mineralization of each compound in soil was quantified as accumulated amounts of 14CO2 during each cycle of biodegradation (Figures 2a and e). Mineralization of 2,4-D was faster than for MCPA, with maximum cumulative mineralization of ∼60% of the initially added 14C after 21 and 26 days for 2,4-D and MCPA, respectively. Maximum mineralization of the second addition of phenoxy acid was reached in 5 and 7 days after amendment for 2,4-D and MCPA, respectively. The shape of the mineralization curve for the first amendment was approximately sigmoidal with a long initial ‘lag’ period showing a very low mineralization rate, followed by rapid mineralization and then a plateau. By contrast, the second mineralization event occurred without noticeable lag. Fitting the observed shapes of the mineralization curves to the degradation models revealed a best fit for the initial amendment of both compounds to the exponential form of the 3/2-order model (Figures 2a and e), while the curves for the second amendment were best fitted to the linear or zero growth form of the 3/2-order model (Figures 2a and e). The choice of best fit for all curves was based on R2-values, F-tests (P<0.05) (data not shown), a general evaluation of how realistic the parameter estimates were (Table 1) and visual inspection of the curve fits.

Phenoxy acid mineralization, gene expression and tfdA gene enumeration in the 2,4-dichlorophenoxyacetic acid (2,4-D) (a–d) and in the 4-chloro-2-methylphenoxyacetic acid (MCPA) (e–h) biodegradation scenarios. Triplicate cumulative mineralization data of 2,4-D and MCPA are shown in (a and e), respectively. In both scenarios, ○ represents the first enrichment, while ▵ represents the second. Lines represent fits to the 3/2-order models. In (b and f), ▪ represents tfdA mRNA quantities during 2,4-D and MCPA biodegradation experiments, respectively, expressed as DNA equivalents. In (c and g) ▴ represents tfdA DNA quantities during the 2,4-D and MCPA experiments, respectively and in (d and h), • represents the ratio of tfdA mRNA (tfdA DNA)−1 in the two experiments. Error bars represent standard errors of the mean for soil triplicates.
Quantitative real-time PCR data
The quantitative gene expression data derived from tfdA mRNA revealed a clear response to presence and absence of 2,4-D and MCPA (Figures 2b and f). No tfdA mRNA was detected in the soil prior to phenoxy acid application, or in negative control microcosms throughout the experiment (data not shown). However, as soon as mineralization of either compound was detected, tfdA mRNA was detected as well. Following this initial mRNA appearance, variable levels of mRNA were detectable throughout the experiment. There was a correlation between levels of tfdA mRNA detected and observed rates of mineralization (Figures 3a and c). For the 2,4-D experiment, tfdA mRNA was detected not before day 13. The abundance increased to the maximum level detected corresponding to ∼104 transcripts per gram soil at day 16, after which it decreased to ∼103 transcripts per gram soil at day 24 (Figures 2b and f). The second amendment with 2,4-D was applied at day 24 and mRNA abundance increased dramatically within just a few hours (Figures 2b and f). Forty-eight hours after the second application, a maximum mRNA level corresponding to ∼105 transcripts per gram soil was observed. In response to the decreasing concentration of 2,4-D, the measure of tfdA transcripts decreased to a minimum after 4 days. With a minor delay in timing of onset, a similar pattern was observed for the MCPA experiment, with the only apparent difference being that the maximum indicated concentration of transcripts after the second application of MCPA was approximately 3 × 105 transcripts per gram soil.

Presence of tfdA mRNA and tfdA genes in relation to the rate of mineralization. In (a and c) ▪ represents the number of tfdA mRNA, while ▴ in (b and d) represents the number of tfdA genes. The panels display copy numbers of mRNA or DNA plotted against the first derivative of time versus percent 14C mineralized for the fitted mineralization curves (refer to Figures 2a and e). Lines display the term of linear regression and R2 values are given. Error bars represent standard errors of the mean for soil triplicates.
Through quantification of tfdA genes based on DNA sequences, there was clear evidence that bacteria harboring tfdA genes increased in abundance during the experiments (Figures 2c and g). Mineralization of the first amendments of 2,4-D and MCPA supported increased abundance of tfdA gene copies from 1 × 104 to 3 × 106 per gram soil, while the second application supported an additional increase from 3 × 106 to 8 × 106 gene copies per gram soil. In terms of growth (assuming that gene copies represent the number of degrading organisms), this corresponds to a 300-fold increase in response to the first application, while the second only supported an additional twofold increase. After mineralization had ceased, we observed decreasing numbers of tfdA genes in both experiments, but not to the background levels observed prior to 2,4-D or MCPA amendment. Contrary to numbers of mRNA transcripts, there was no significant correlation between indicated numbers of gene copies and rates of mineralization (Figures 3b and d).
Melting curve profile analysis
Melting curve profile analyses were performed on real-time qPCR products from soil extracts along with type strains containing tfdA class I and III DNA (Figure 4). Melting curve profiles represent a separate, confirmatory assay in real-time PCR analyses using the fluorophore SYBR Green and are used to validate the presence and quality of a proper PCR product. Depending on the nucleotide sequence of the PCR product, different products will exhibit different melting curve profiles. As the class I, II and III tfdA PCR products exhibit different melting curves, we were able to use this analysis to establish the presence of these three gene types. Class II tfdA genes were not detected in any of the soil extracts and therefore, in order to simplify Figure 4, the type strain for this gene was not included as a reference.

Melting curve profiles of real-time PCR products using DNA and cDNA as template. In panel a, melting for class I ( ) and class III (
) and class III ( ) positive control organisms are shown. Representative melting curves from the 2,4-D soil scenario are shown in panels b and d for DNA and cDNA, respectively. Representative melting curves from the MCPA soil scenario are shown in panels c and e for DNA and cDNA, respectively. In panels b–e,
) positive control organisms are shown. Representative melting curves from the 2,4-D soil scenario are shown in panels b and d for DNA and cDNA, respectively. Representative melting curves from the MCPA soil scenario are shown in panels c and e for DNA and cDNA, respectively. In panels b–e,  ,
,  and
and  represent the melting curves of PCR products from soil samples, sampled at day 0, an early stage and a late stage of biodegradation, respectively. The panels display the negative first derivative of temperature versus relative fluorescence units (−d(RFU)/dT) plotted against temperature.
represent the melting curves of PCR products from soil samples, sampled at day 0, an early stage and a late stage of biodegradation, respectively. The panels display the negative first derivative of temperature versus relative fluorescence units (−d(RFU)/dT) plotted against temperature.
On the basis of melting curves of qPCR products from DNA samples, the soil contained a relatively low, but detectable, background population (at day 0, prior to soil amendments) of bacteria harboring the class I tfdA gene (Figures 4a and b). Still, based on melting curves from DNA-based qPCR products, it is clear that a peak with a similar temperature profile as the class III tfdA gene from the positive control emerged as time passed. As no class III tfdA gene copies were detected in the soil prior to pesticide application, this indicates that organisms harboring this gene substantially increased in abundance during degradation of 2,4-D (Figure 4a) as well as MCPA (Figure 4b). While melting curve profiles can probably not be regarded as precisely quantitative, the general pattern of peaks observed suggests that the class I:III gene ratio remained in favor of class I genes in the 2,4-D experiment, while the same ratio shifted to favor class III genes in the MCPA experiment.
Analysis of the melting curve profiles for mRNA-based (that is, cDNA) PCR products (Figures 4c and d) reveals an interesting pattern. In the soil, prior to pesticide application (at day 0), there were no detectable tfdA transcripts. As degradation occurred, both class I and III tfdA genes were expressed in the 2,4-D experiment (Figure 4c), while only class III mRNA was detected in the MCPA experiment (Figure 4d). Taking into account the entire set of melting curve profiles (some data not shown), a clear trend was that the peak representing tfdA class III was relatively small compared to the peak representing class I tfdA genes during the early stage of the 2,4-D experiment (days 8–14). During the later stages (days 16 to the end of the experiment), the two peaks became more comparable. A similar trend was observed at the DNA level (Figure 4a), but the two peaks never became equal in size. In contrast, during the MCPA experiment the DNA-based class I tfdA peak evolved from being dominant in the beginning of the experiment (day 0–7) to being negligible after the first major mineralization and growth event had occurred (day 15 to the end of the experiment). At the mRNA level, only the tfdA class III peak was detectable, which suggests that only class III genes and organisms proliferated.
DGGE on functional genes
PCR for DGGE analysis was performed using the same DNA extracts for template as were used for the melting curve analysis described above. In general, the patterns observed from the DGGE gels are highly comparable to those described in the melting curve section above. For the 2,4-D experiment (Figure 5a), analyses at the DNA level indicated a strong increase in tfdA class III genes, going from undetectable at day 0 to a very strong band at later time points. The increased intensity observed in the band representing tfdA class I genes is of lower magnitude than observed for the class III gene. At the mRNA level, the band intensities representing class III transcripts were much more pronounced than those representing class I transcripts. For the MCPA experiment, the DNA-based assay showed a strong increase in class III genes, while the class I genes were more or less unchanged throughout the experiment (Figure 5b). The mRNA analyses showed that class III genes were strongly transcribed during MCPA degradation, while no transcription was detected for class I genes at all. For the MCPA experiment, the area where class I transcripts would have appeared (Figure 5b, arrows 8 and 9) appeared to be completely clear, while the comparable area in the 2,4-D experiment (Figure 5a, arrows 4 and 5) showed a low-intensity signal. The bands denoted with arrows were excised from the gels and sequenced (data not shown), which indicated that bands 1, 2, 6 and 7 had >99% homology to class III tfdA genes, while bands 3, 4 and 5 had >99% homology to class I tfdA genes. As expected, areas 8 and 9 gave no result.

tfdA-denaturing gradient gel electrophoresis (DGGE) using the same DNA and cDNA for template in PCR reactions as for the melting curves (Figure 4). Arrows and numbers denote bands that were excised from the gel and subjected to DNA sequence analysis. Bands 1, 2, 6 and 7 had >99% homology to class III tfdA genes, bands 3, 4 and 5 had >99% homology to class I tfdA genes and the areas indicated by 8 and 9 gave no sequencing result. (a) 2,4-dichlorophenoxyacetic acid (2,4-D) soil scenario and (b) 4-chloro-2-methylphenoxyacetic acid (MCPA) soil scenario. M lane is a marker that consists of tfdA PCR products from class I, II and III represented by Cupriavidus necator JMP134, Burkholderia sp. RASC and an unclassified strain, respectively.
Discussion
The study reported herein examined the dynamics of functional gene expression and the diversity present in functional gene transcripts in a natural soil during biodegradation of phenoxyacetic acids, and demonstrates that quantitative and qualitative gene expression analyses can indeed be achieved for indigenous bacterial populations in a natural soil. Transcriptional analyses on indigenous populations in soil have been a desirable yet elusive goal of microbial ecologists for the past 2 decades. Consequently, only a few studies have been published investigating gene transcription directly in soil (Saleh-Lakha et al., 2005). Recently, improvements in extraction methods, especially for mRNA, and downstream quantitative and qualitative detection assays have added tractability to transcriptional analyses in soil (Bælum et al., 2006; Nicolaisen et al. 2008).
Functional diversity within 2,4-D-degrading isolates has been studied in great detail (Fulthorpe et al., 1995; Vallaeys et al., 1996; Kamagata et al., 1998; Kitagawa et al., 2002), but information on dynamics and diversity among the potential degraders (that is, organisms encoding tfdA and tfdA-like genes) and the active degraders (that is, organisms actually expressing tfdA or tfdA-like genes) in situ in soil is still rather scarce. Bælum et al. (2006) studied growth and functional diversity among potential degraders in one soil. Based on DNA analyses, it was suggested that class III tfdA-harboring organisms were responsible for the majority of MCPA degradation. In the current report we provide additional support to that conclusion and extend on those studies in several ways, namely: (1) using mRNA analyses on indigenous populations to demonstrate which tfdA classes are being expressed in soil, (2) including the structurally related compound 2,4-D for contrast and comparison and (3) by extending the generality of the prior finding using a different soil.
By analyzing the diversity of tfdA genes based on the cDNA pool (that is, from mRNA) rather than on the soil DNA pool, important information was obtained on which classes of tfdA genes were actually and actively expressed during biodegradation. Melting curve profiles for the real-time PCR products (Figures 4a–d) indicated that the canonical type of phenoxyacetic acid-degrading bacteria harboring the class I tfdA gene were not primarily responsible for MCPA degradation in the soil tested. Class I tfdA genes were present at a detectable background level in the soil, but only the 2,4-D amendment resulted in increased copy number and expression of class I tfdA genes. Class III tfdA genes, however, were not detected prior to treatment, but increased in number and were actively expressed in the presence of both 2,4-D and MCPA. Interestingly, this observation was made despite the fact that numerous pure cultures isolated on 2,4-D as carbon source have been shown to degrade MCPA, even though they apparently only possess the tfdA class I gene (Smejkal et al., 2001). This suggests that pure culture studies may bias our understanding of what is actually occurring in situ in natural environments. In contrast, biodegradation of 2,4-D seems to be performed by bacteria representing a larger functional diversity (that is, both class I and class III bacteria).
DGGE was used to support the melting curve analysis and consistent patterns were revealed. Further, bands excised from the DGGE gels established that the PCR products represented by the peaks in the melting curve profiles indeed represented the suggested tfdA classes. Neither the melting curve analysis nor the DGGE analysis can be regarded as reliably quantitative, meaning that the precise dynamics between class I and III harboring organisms in the 2,4-D experiment are still uncertain. Nonetheless, based on these two analyses, it was clear that only class III tfdA genes were expressed and quantified at detectable levels during degradation of MCPA. As reported previously for a different soil (Bælum et al., 2006) and confirmed above, this indicates that MCPA degradation in these natural soil environments is exclusively mediated by organisms carrying the class III tfdA gene. In a recent study by Zakaria et al. (2007), similar results were obtained for another structurally related phenoxy acid compound, mecoprop, where only class III tfdA gene sequences were obtained from a tfdA gene library constructed from mecoprop enrichments of soil microbial communities.
In the 2,4-D experiments, different time points were chosen for the DNA and cDNA utilized for the melting curve and DGGE analyses. This derives from the fact that our goal was to elucidate the dynamics between class I and III tfdA genes in soil. For the DNA analysis, day 16 was chosen because it was the first time point where the class III tfdA gene was detected in the melting curves, while day 24 was chosen to depict the dynamics after one complete mineralization event. The cDNA melting curves exemplify how mRNA analyses are more sensitive and precise than DNA analyses since the class III tfdA gene was detected earlier (at day 8). Day 16 was chosen to represent a time point when a high mineralization rate and high concentration of mRNA were apparent. For the MCPA experiment in which only class III genes were detected, day 7 was chosen because neither class III tfdA genes nor mRNA were detected at earlier times, and day 36 is presented for its high concentration of tfdA mRNA and gene dosage.
Transcription of tfdA genes showed a clear response to the introduction of phenoxyacetic acid compounds as the relative number of transcripts increased in response to amendment and subsequently decreased after the compounds were degraded. These expression patterns resemble the classical patterns of functional gene expression observed for pure cultures (Dennis et al., 2003; Devers et al., 2004). However, due to the prior intractability of mRNA studies in situ, such patterns have never been demonstrated for functional genes of indigenous bacteria in complex soil environments. The relative number of transcripts per tfdA gene in the soil was determined during 2,4-D and MCPA degradation scenarios. The term ‘DNA equivalents’ was used to describe the ratio of mRNA:DNA that was detected for each tfdA class. We believe this general approach represents the most reliable normalization protocol available to quantify gene expression in situ. Alternatively, quantification of mRNA from functional genes might be normalized to transcripts of a so-called ‘housekeeping gene’ that is constitutively and evenly transcribed during all growth phases and states of metabolic activity (Eleaume and Jabbouri, 2004; Johnson et al., 2005). However, to our knowledge, no such gene has been suggested across all of prokaryotic diversity and such normalization would require extensive a priori knowledge of the suites of genes present in natural soil microorganisms, which is currently not available.
As in our previous study (Bælum et al., 2006), quantitative analyses based on extracted DNA indicated growth of bacteria harboring tfdA genes during 2,4-D and MCPA biodegradation in soil. The ∼3 × 106 gene copies per gram soil detected in both degradation scenarios (refer to Figure 2) is also consistent with that previous study. New dimensions included in the present study include the use of a different soil, the inclusion of the 2,4-D biodegradation scenario, and the second cycle of enrichment event for both MCPA and 2,4-D. In terms of tfdA gene copy numbers, the 2,4-D biodegradation scenario revealed a similar pattern to the MCPA biodegradation scenario, except that the degradation of MCPA was somewhat slower and therefore the increase in gene copy numbers was delayed relative to the 2,4-D scenario. Both phenoxy acid compounds ultimately produced similar increases in gene copy numbers for both enrichment events, with the first enrichment producing a ∼300-fold increase in gene copy number, while the second resulted in only a ∼2-fold additional increase. This was as expected since the 3 × 106 copies resulting from the initial event represent significant potential for rapid biodegradation when the second addition was made and thus little additional growth was required or supported by the second addition. This behavior was manifested in the mineralization curves as well, with the curves for the first enrichment being best fitted to the exponential growth form of the 3/2-order model indicating growth during degradation (Brunner and Focht, 1984), while the second enrichment was best fitted to the linear growth form of the 3/2-order model indicating no or linear growth during degradation.
As DNA (in the form of bacterial genomes) does not decay unless cells die and degrade, while mRNA in viable cells is expected to turn over fairly rapidly following depletion of the substrate whose biodegradation it encodes, the lack of correlation between numbers of tfdA genes and rates of mineralization was expected. When mineralization had ceased after the first round of enrichment, tfdA gene DNA sequences remained in the soil in high numbers even at low mineralization rates (Figure 3). The correlation between numbers of tfdA mRNA transcripts detected and measured rates of mineralization, on the other hand, is a manifestation of the rapid decay of mRNA and its absence during low rates of mineralization. It is important to remember that the presence of a gene is an indicator of potential gene expression, not an actual measure of it. This clearly and emphatically demonstrates why measurement of mRNA (where tractable) is a far better indicator of in situ metabolic activity than measurement of the number of copies of the gene that are present in an environmental sample.
Environmental studies based on functional gene transcripts are potentially complicated by several critical factors. For one, many processes proceed slowly in the environment due to a variety of environmental parameters and limiting factors and therefore require only low amounts of mRNA. In addition, mRNA in active biological matrices is extremely labile, with reported half-lives being as short as just a few minutes (Rauhut and Klug, 1999; Selinger et al., 2003). Further, nucleic acids, including mRNA, are strongly sorbed to soil and sediment components (Chamier et al., 1993), which can significantly affect extraction efficiency. Finally, extraction efficiencies of mRNA are difficult to assess as the use of internal standards for normalization of mRNA extraction efficiency is currently not possible for prokaryotic transcripts in environmental matrices. A carefully optimized extraction protocol (Nicolaisen et al., 2008) is obviously of major importance when studying environmental mRNA, but the downstream quantitative and qualitative detection assays must also be considered to be at least of equal importance. In the current study, we used random hexamer primers in the RT reaction prior to quantification by real-time PCR to ensure that maximum diversity of extracted mRNA was captured in the resultant cDNA product. Although gene-specific primers might potentially be considered to be more effective, or at least more direct, enabling production of specific cDNA molecules, our strategy produced cDNA representing the fully expressed soil genome. Thus, the cDNA pool from these samples could be used to analyze transcripts from other genes as well. In addition, Nicolaisen et al. (2008) found that nonspecific amplification products were formed during subsequent PCR when using gene specific primers to produce cDNA from total community mRNA.
We note that the effective application of mRNA-based methodologies in the near term will be limited to genes that have been studied in detail. The sequence of the gene of interest must be known and PCR primers must be designed to cover its potential diversity in the microbial community. Presumably, advances in comparative genomics will help alleviate this limitation in years to come. Even with progress in that area, mRNA-based quantification of gene expression in natural soils may still be hampered by low sensitivity. Hence, it is important that the genes of interest are actually expressed to a detectable level, and studies of slow soil processes with correspondingly low transcription levels may always be limited.
In conclusion, the data reported herein provides the first example of an in-depth quantitative analysis of functional gene expression in natural soil samples not seeded with microbes or mRNA. The study demonstrates that analyzing soil microbial communities for a specific metabolic activity based on DNA sequences may be biased by detection of nonactive populations encoding homologous genes, but not actively expressing them. Further, we believe that the ability to detect and quantify functional gene transcripts in a complex environmental matrix will become an important tool for microbial ecologists striving to link phylogeny to ecological function and should help to improve our understanding of microbial processes in general.
References
Andersen SM, Hertz PB, Holst T, Bossi R, Jacobsen CS . (2001). Mineralization studies of 14C-labelled metsulfuron-methyl, tribenuronmethyl, chlorsulfuron, and trifensulfuron-methyl in one Danish soil and groundwater sediment profile. Chemosphere 45: 775–782.
Bælum J, Henriksen T, Hansen HCB, Jacobsen CS . (2006). Degradation of 4-chloro-2-methylphenoxyacetic acid in top- and subsoil is quantitatively linked to the class III tfdA gene. Appl Environ Microbiol 72: 1476–1486.
Brunner W, Focht DD . (1984). Deterministic three-half-order kinetic model for microbial degradation of added carbon substrates in soil. Appl Environ Microbiol 47: 167–172.
Chamier B, Lorenz MG, Wackernagel W . (1993). Natural transformation of Acinetobacter calcoaceticus by plasmid DNA adsorbed on sand and groundwater aquifer material. Appl Environ Microbiol 59: 1662–1667.
Dennis P, Edwards EA, Liss SN, Fulthorpe R . (2003). Monitoring gene expression in mixed microbial communities by using DNA microarrays. Appl Environ Microbiol 69: 769–778.
Deutscher MP . (2006). Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucl Acids Res 34: 659–666.
Devers M, Soulas G, Martin-Laurent F . (2004). Real-time reverse transcription PCR analysis of expression of atrazine catabolism genes in two bacterial strains isolated from soil. J Microbiol Methods 56: 3–15.
Don RH, Weightman AJ, Knackmuss HJ, Timmis KN . (1985). Transposon mutagenesis and cloning analysis of the pathways for degradation of 2,4-dichlorophenoxyacetic acid and 3-chlorobenzoatein Alcaligenes eutrophus JMP134(pJP4). J Bacteriol 161: 85–90.
Eleaume H, Jabbouri S . (2004). Comparison of two standardisation methods in real-time quantitative RT-PCR to follow Staphylococcus aureus genes expression during in vitro growth. J Microbiol Methods 59: 363–370.
Fukimori F, Hausinger RP . (1993). Purification and characterization of 2,4-dichlorophenoxyacetate alpha-ketogluterate dioxygenase. J Biol Chem 268: 24311–24317.
Fulthorpe RR, McGowan C, Maltseva OV, Holben WE, Tiedje JM . (1995). 2,4-Dichlorophenoxyacetic acid-degrading bacteria contain mosaics of catabolic genes. Appl Environ Microbiol 61: 3274–3281.
Han JI, Semrau JD . (2004). Quantification of gene expression in methanotrophs by competitive reverse transcription-polymerase chain reaction. Environ Microbiol 6: 388–399.
Itoh K, Kanda R, Momoda Y, Sumita Y, Kamagata Y, Suyama K et al. (2000). Presence of 2,4-D-catabolizing bacteria in a Japanese arable soil that belong to BANA (Bradyrhizobium-Agromonas-Nitrobacter-Afipia) cluster in α-Proteobacteria. Microb Environ 15: 113–117.
Jacobsen CS, Holben WE . (2007). Quantification of mRNA in Salmonella sp. seeded soil and chicken manure using magnetic capture hybridization RT-PCR. J Microbiol Methods 69: 315–321.
Johnson DR, Lee PKH, Holmes VF, Alvares-Cohen L . (2005). An internal reference technique for accurately quantifying specific mRNAs by real time PCR with application to the tceA reductive dehalogenase gene. Appl Environ Microbiol 71: 3866–3871.
Kamagata Y, Fulthorpe RR, Tamura K, Takami H, Forney LJ, Tiedje JM . (1998). Pristine environments harbour a new group of oligotrophic 2,4-dichlorophenoxy acid-degrading bacteria. Appl Environ Microbiol 63: 2266–2272.
Kitagawa W, Takami S, Miyauchi K, Masai E, Kamagata Y, Tiedje JM et al. (2002). Novel 2,4-dichlorophenoxyacetic acid degradation genes from oligotrophic Bradyrhizobium sp. strain HW13 isolated from a pristine environment. J Bacteriol 184: 509–518.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW et al. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soil. Nature 442: 806–809.
Lindahl T . (1993). Instability and decay of the primary structure of DNA. Nature 362: 709–715.
McGowan C, Fulthorpe R, Wright A, Tiedje JM . (1998). Evidence for interspecies gene transfer in the evolution of 2,4-dichlorophenoxyacetic acid degraders. Appl Environ Microbiol 64: 4089–4092.
Mortensen SK, Jacobsen CS . (2004). Influence of frozen storage on herbicide degradation capacity in surface and subsurface sandy soils. Environ Sci Technol 38: 6625–6632.
Nicolaisen MH, Bælum J, Jacobsen CS, Sørensen J . (2008). Transcription dynamics of the functional tfdA gene during MCPA herbicide degradation by Cupriavidus necator AEO106 (pRO101) in agricultural soil. Environ Microbiol 10: 571–579.
Nogales B, Timmis KN, Nedwell DB, Osborn AM . (2002). Detection and diversity of expressed denitrification genes in estuarine sediments after reverse transcription-PCR amplification from mRNA. Appl Environ Microbiol 68: 5017–5025.
Pemberton JM, Corney B, Don RH . (1979). Evolution and spread of pesticide degrading ability among soil microorganisms, In: Timmis KN, Pühler A (eds). Plasmids of Medical Environmental and Commercial Importance. Elsevier: Amsterdam, The Netherlands, pp 287–299.
Rauhut R, Klug G . (1999). mRNA degradation in bacteria. FEMS Microbiol Rev 23: 353–370.
Saleh-Lakha S, Miller M, Campbell RG, Schneider K, Elahimanesh P, Hart MM et al. (2005). Microbial gene expression in soil: methods, applications and challenges. J Microbiol Methods 63: 1–19.
Selinger DW, Saxena RM, Cheung KJ, Church GM, Rosenow C . (2003). Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res 13: 216–223.
Smejkal CW, Vallaeys T, Burton SK, Lappin-Scott HM . (2001). Substrate specificity of chlorophenoxyalkanoic acid-degrading bacteria is not dependent upon phylogenetically related tfdA gene types. Biol Fertil Soils 33: 507–513.
Stanier RY, Palleroni NJ, Doudoroff M . (1966). Aerobic Pseudomonads—a taxonomic study. J Gen Microbiol 43: 159–271.
Tonso NL, Matheson VG, Holben WE . (1994). Polyphasic characterization of a suite of bacterial isolates capable of degrading 2,4-D. Microb Ecol 30: 3–24.
Vallaeys T, Fulthorpe RR, Wright AM, Soulas G . (1996). The metabolic pathway of 2,4-dichlorophenoxyacetic acid degradation involves different families of tfdA and tfdB genes according to PCR-RFLP analysis. FEMS Microbiol Ecol 20: 163–172.
Zakaria D, Lappin-Scott H, Burton S, Whitby C . (2007). Bacterial diversity in soil enrichment cultures amended with 2 (2-methyl-4-chlorophenoxy) propionic acid (mecoprop). Environ Microbiol 9: 2575–2587.
Acknowledgements
We thank The Danish Research Council for Technology and Production Sciences for economic support of the project ‘Agricultural practise, microbial activity and pesticide leaching’ (FTP no. 274-05-0199). We also thank the Villum Kann Rasmussen Foundation for its support to the Center of Environmental and Agricultural Microbiology (CREAM). Moreover, we thank Mette Munch for technical laboratory support and Jacob Clemmensen for mathematical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bælum, J., Nicolaisen, M., Holben, W. et al. Direct analysis of tfdA gene expression by indigenous bacteria in phenoxy acid amended agricultural soil. ISME J 2, 677–687 (2008). https://doi.org/10.1038/ismej.2008.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.21
Keywords
This article is cited by
-
Bioaugmentation has temporary effect on anaerobic pesticide biodegradation in simulated groundwater systems
Biodegradation (2024)
-
Dosage concentration and pulsing frequency affect the degradation efficiency in simulated bacterial polycyclic aromatic hydrocarbon-degrading cultures
Environmental Science and Pollution Research (2023)
-
Potential and limitations for monitoring of pesticide biodegradation at trace concentrations in water and soil
World Journal of Microbiology and Biotechnology (2022)
-
Modified zeolite-supported biofilm in service of pesticide biodegradation
Environmental Science and Pollution Research (2021)
-
Structural and functional shifts of soil prokaryotic community due to Eucalyptus plantation and rotation phase
Scientific Reports (2020)
