
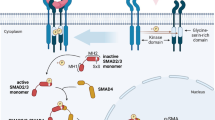
Abstract
Currently, surgical intervention is the only efficacious treatment for Peyronie's disease (PD), a fibromatosis of the tunica albuginea of the penis. Therapies based on the molecular pathways for this disease could provide alternatives to surgical treatment but only recently has the pathophysiology of the Peyronie's disease plaque been investigated at the molecular level. In this review, we examine the current knowledge of gene expression in the PD plaque and the relationship of PD with other fibrotic conditions such as Dupytren's disease. TGFβ1, along with other growth factors, pro-fibrotic genes, and collagen, are expressed in fibroblasts and myofibroblasts. Myofibroblasts are normally involved in wound contracture and largely eliminated via apoptosis during the late stages of wound remodeling. In the PD plaque, however, these cells persist and may play an important role in the PD plaque fibrosis. The expression levels of TGFβ1 and pro- and anti-fibrotic gene products, along with the nitric oxide/reactive oxygen species (NO/ROS) ratio in the tunica albuginea, appear to be essential for the formation and progression of the PD plaque and effect the expression of multiple genes. This can be assessed with the recently developed DNA-based chip arrays and results with the PD plaque have been encouraging. OSF-1 (osteoblast recruitment), MCP-1 (macrophage recruitment), procollagenase IV (collagenase degradation), and other fibrotic genes have been identified as being possible candidate regulatory genes. Finally, possible therapeutic avenues for gene-based therapy in the treatment of PD are discussed that may eventually reduce the need for surgical intervention.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 8 print issues and online access
$259.00 per year
only $32.38 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
References
Jordan GH, Schlossberg SM, Devine CJ . Surgery of the penis and urethra In: Walsh PC, Retik AB, Vaughan ED, Wein AJ (eds) Campbell's urology 7th Edition WB Saunders: Philadelphia 1998 pp 3376–3394
Levine LA . Advances in the medical therapy of Peyronie's disease: a brief review Int J Impot Res 1998 10: 123–124
Noss MB, Day NS, Christ GJ, Melman A . The genetics and immunology of Peyronie's disease Int J Impot Res 2000 4(Suppl): S127–132
Hellstrom WJ, Bivalacqua TJ . Peyronie's disease: etiology, medical, and surgical therapy J Androl 2000 21: 347–354
Gholami SS, Lue TF . Peyronie's disease Urol Clin North Am 2001 28: 377–390
Rhoden EL et al. Prevalence of Peyronie's disease in men over 50 years old J Urol 2001 165: 200 (#827)
McVary KT . Peyronie's disease In: Plenary Session 3, American Urological Association Annual Meeting 2001
Davis CJ . The microscopic pathology of Peyronie's disease J Urol 1997 157: 282–284
Devine CJ Jr, Somers KD, Jordan SG, Schlossberg SM . Proposal: trauma as the cause of the Peyronie's lesion J Urol 1997 157: 285–290
Jarow JP, Lowe FC . Penile trauma: an etiologic factor in Peyronie's disease and erectile dysfunction J Urol 1997 158: 1388–1390
Ehrlich HP . Scar contracture: cellular and connective tissue aspects in Peyronie's disease J Urol 1997 157: 316–319
Van de Water L . Mechanisms by which fibrin and fibronectin appear in healing wounds: implications for Peyronie's disease J Urol 1997 157: 306–310
Somers KD, Dawson DM . Fibrin deposition in Peyronie's disease plaque J Urol 1997 157: 311–315
Diegelmann RF . Cellular and biochemical aspects of normal and abnormal wound healing: an overview J Urol 1997 157: 298–302
Martnez-Hernández A . Repair, regeneration and fibrosis Pathology 2nd Edition, Chapter 3 JB Lippincott: Philadelphia 1994 p 69
Singer AJ, Clark RAF . Cutaneous wound healing N Engl J Med 1999 341: 738–746
Clark AF . Wound repair: overview and general considerations In: Clark RAF (ed) The molecular and cellular biology of wound repair 2nd Edition Plenum Press: New York 1996 pp 3–50
Klahr S, Morrisey JJ . The role of growth, cytokines and vasoactive compounds in obstructive nephropathy Seminars Nephrol 1998 18: 622–632
El-Sakka AI et al. An animal model of Peyronie's-like condition associated with an increase of transforming growth factor beta mRNA and protein expression J Urol 1997 158: 2284–2290
El-Sakka AI et al. Histological and ultrastructural alterations in an animal model of Peyronie's disease British J Urol 1998 81: 445–452
El-Sakka AI et al. The effect of surgical trauma on rat tunica albuginea J Urol 1998 159: 1700–1707
Hirano D et al. Electron microscopic study of the penile plaques and adjacent corpora cavernosa in Peyronie's disease Int J Urol 1997 4: 274–278
Akkus E et al. Structural alterations in the tunica albuginea of the penis: impact of Peyronie's disease, ageing and impotence Br J Urol 1997 79: 47–53
Gentile V et al. Ultrastructural and immunohistochemical characterization of the tunica albuginea in Peyronie's disease and veno-occlusive dysfunction J Androl 1996 17: 96–103
Brock G et al. The anatomy of the tunica albuginea in the normal penis and Peyronie's disease J Urol 1997 157: 276–281
El-Sakka AI et al. The effects of colchicine on a Peyronie's-like condition in an animal model J Urol 1999 161: 1980–1983
Bivalacqua TJ et al. A rat model of Peyronie's disease associated with a decrease in erectile activity and an increase in inducible nitric oxide synthase protein expression J Urol 2000 163: 1992–1998
Bivalacqua TJ et al. Evaluation of nitric oxide synthase and arginase in the induction of a Peyronie's-like condition in the rat J Androl 2001 22: 497–508
Ferrini MG et al. Antifibrotic role of inducible nitric oxide synthase (iNOS) Nitric Oxide 2002 6: 1–12
El-Sakka AI et al. Peyronie's disease is associated with an increase in transforming growth factor-beta protein expression J Urol 1997 158: 1391–1394
Powel DW et al. Myofibroblasts. Paracrine cells important in health and disease Am J Physiol 1999 277: C1–C19
Gabbiani G . The cellular derivation and the life span of the myofibroblasts Path Res Pract 1996 192: 701–711
Desmoulière A . Factors influencing myofibroblasts differentiation during wound healing and fibrosis Cell Biol Int 1995 19: 471–476
Walker GA, Guerrero IA, Leinwand LA . Myofibroblasts: Molecular crossdressers Curr Top Devel Biol 2001 51: 91–107
Tomasek JJ, Vaugham MB, Haaksma CJ . Cellular structure and biology of Dupuytren's disease Hand Clinics 1999 15: 21–34
Somers KD et al. Cell culture of Peyronie's disease plaque and normal penis tissue J Urol 1982 127: 585–588
Mulhall JP, Thorn J, Lubrano T, Shankey TV . Basic fibroblast growth factor expression in Peyronie's disease J Urol 2001 165: 419–423
Vernet D et al. Differentiation phenotype of myofibroblasts from the penile tunica albuginea J Urol 2001 165: 200 (#829)
Wahl SM . Inflammation and growth factors J Urol 1997 157: 303–305
Badalamante MA et al. The role of transforming growth factor beta in Dupuytren's disease J Hand Surg 1996 21A: 210–215
Roberts AB, Sporn MB . Transforming growth factor β In: Clark RAF, ed The molecular and cellular biology of wound repair 2nd Edition Plenum Press: New York 1996 pp 275–310
Poli G, Parola M . Oxidative damage and fibrogenesis Free Radic Biol Med 1997 22: 287–305
Poli G . Pathogenesis of liver fibrosis: role of oxidative stress Mol Aspects Med 2000 21: 49–98
Casini A et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide Hepatology 1997 25: 361–367
Muriel P . Nitric oxide protection of rat liver from lipid peroxidation, collagen accumulation, and liver damage induced by carbon tetrachloride Biochem Pharmacol 1998 56: 773–779
Geller DA, Billiar TR . Molecular biology of nitric oxide synthases Cancer Metast Rev 1998 17: 7–23
Nathan C . Inducible nitric oxide synthase: What difference does it make? J Clin Invest 1997 100: 2417–2423
Beckmann JS, Koppenol WH . Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly Am J Physiol 1996 271: C1424–C1437
Hamilton CA et al. Superoxide excess in hypertension and aging. A common cause of endothelial dysfunction Hypertension 2001 37: 529–534
Wink D et al. Antioxidant effects of nitric oxide Meth Enzymol 1999 301: 413–424
Patel RP et al. Cell signaling by reactive nitrogen and oxygen species in atherosclerosis Free Radic Biol Med 2000 28: 1780–1794
Higuchi H, Granger DN, Saito H, Kurose I . Assay of antioxidant and anti-inflammatory activity of nitric oxide in vivo. Meth Enzymol 1999 301: 424–436
Foresti R et al. Peroxynitrite induces haemoxygenase-1 in vascular endothelial cells: a link to apoptosis Br Biochem J 1999 339: 729–736
Franklin TJ . Therapeutic approaches to organ fibrosis Int J Biochem Cell Biol 1997 29: 79–89
Campbell SE, Katwa LC . Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts J Mol Cell Cardiol 1997 29: 1947–1958
Dussaule JC et al. Mechanisms mediating the renal profibrotic actions of vasoactive peptides in transgenic mice J Am Soc Nephrol Suppl 2000 16: S124–128
Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC . Nitric oxide inhibition induces early activation of type I collagen gene in renal resistance vessels and glomeruli in transgenic mice. Role of endothelin J Clin Invest 1998 101: 2780–2789
Tharaux PL et al. Vascular endothelin-1 gene expression and synthesis and effect on renal type I collagen synthesis and nephroangiosclerosis during nitric oxide syntahse inhibition in rats Circulation 1999 99: 2185–2191
Boffa JJ et al. Angiotensin II activates collagen I gene in the renal vasculature of transgenic mice during inhibition of nitric oxide synthesis. Evidence for an endothelium-mediated mechanism Circulation 1999 100: 1901–1908
Rizvi MA, Myers PR . Nitric oxide modulates basal and endothelin-induced coronary artery vascular smooth muscle cell proliferation and collagen levels J Mol Cell Cardiol 1997 29: 1779–1789
Weber KT, Sun Y, Katwa LC . Myofibroblasts and local angiotensin II in rat cardiac tissue repair Int J Biochem Cell Biol 1997 29: 31–42
Gonzalez-Cadavid NF, Ignarro, Rajfer J . Nitric oxide and cyclic GMIP in the penis Mol Urol 1999 3: 51–59
Burnett AL . Nitric oxide in the penis: physiology and pathology J Urol 1997 157: 320–324
Schaffer MR et al. Nitric oxide, an autocrine regulator of wound fibroblast synthetic function J Immunol 1997 158: 2375–2381
Schaffer MR et al. Nitric oxide regulates wound healing J Surg Res 1996 63: 237–240
Hogaboam CM et al. Collagen deposition in a non-fibrotic lung granuloma model after nitric oxide inhibition Am J Pathol 1998 153: 1861–1872
Schaffer MR et al. Diabetes-impaired healing and reduced wound nitric oxide synthesis: a possible pathophysiologic correlation Surgery 1997 121: 513–519
Thornton FJ et al. Enhanced collagen accumulation following direct transfection of the inducible nitric oxide synthase gene in cutaneous wounds Biochem Biophys Res Commun 1998 246: 654–659
Yamasaki K et al. Reversal of impaired wound repair in iNOS-deficient mice by topical adenoviral-mediated iNOS gene transfer Am J Physiol 1998 101: 967–971
Chung HT et al. Antisense transforming growth factor-beta 1 in wound healing Antis Nucl Acid Drug Devel 1997 7: 257–261
Cao M et al. Nitric oxide inhibits the synthesis of type-II collagen without altering Co12A1 mRNA abundance: prolyl hydroxylase as a possible target Biochem J 1997 324: 305–310
Kolpakov V, Gordon D, Kulik TJ . Nitric oxide-generating compounds inhibit total protein and collagen synthesis in cultured vascular smooth muscle cells Circul Res 1995 76: 305–309
Catani MV et al. Inhibition of clotting factor XIII activity by nitric oxide Biochem Biophys Res Comm 1998 249: 275–278
Dambisya YM et al. A thromboelastography study on the in vitro effects of L-arginine and L-NG-nitro arginine methyl ester on human whole blood coagulation and fibrinolysis Blood Coagul Fibrinol 1996 7: 678–683
Craven PA et al. Nitric oxide inhibition of transforming growth factor-beta and collagen synthesis in mesangial cells Diabetes 1997 46: 671–681
Takemoto M et al. Chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade: effects on cardiovascular remodeling in rats induced by the long-term blockade of nitric oxide synthesis Hypertension 1997 30: 1621–1627
Babal P, Pechanova O, Bernatova I, Stvrtina S . Chronic inhibition of NO synthesis produces myocardial fibrosis and arterial media hyperplasia Histol Histopath 1997 12: 623–629
Numaguchi K et al. Chronic inhibition of nitric oxide synthesis causes coronary microvascular remodeling in rats Hypertension 1995 26: 957–962
Ikeda K, Nara Y, Tagami M, Yamori Y . Nitric oxide deficiency induces myocardial infarction in hypercholesterolemic stroke-prone spontaneously hypertensive rats Clin Exptl Pharmacol Physiol 1997 24: 344–348
Moreno H Jr et al. Chronic nitric oxide inhibition as a model of hypertensive heart muscle disease Basic Res Cardiol 1996 91: 248–255
Morrissey JJ, Ishidoya S, McCracken R, Klahr S . Nitric oxide generation ameliorates the tubulointerstitial fibrosis of obstructive nephropathy J Am Soc Nephrol 1996 70: 2202–2212
Kelley TJ, Drumm ML . Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells J Clin Invest 1998 102: 1200–1207
Ferrini M et al. Aging-related increased expression of inducible nitric oxide synthase and cytotoxicity markers in rat hypothalamic regions associated with male reproductive function Neuroendocrinology 2001 74: 1–11
Lue YH et al. Decrease in induction of germ cell apoptosis by a single heat exposure in iNOS deficient mice 82nd Endocr Soc Meet 2000 #1384
Ferrini M et al. Aging-related expression of inducible nitric oxide sunthase (iNOS) and cytotoxicity markers in the rat penis Biol Reprod 2001 64: 974–982
Boger RH et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia Circulation 1998 98: 1842–1847
Kelley TJ, Drumm ML . Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells J Clin Invest 1998 102: 1200–1207
Hochberg D et al. Interstitial fibrosis of unilateral ureteral obstruction is exacerbated in kidneys of mice lacking the gene for inducible nitric oxide synthase Lab Invest 2000 80: 1721–1728
Vernet D et al. Age-related increase in germ cell apoptosis in male Brown-Norway (BN) rats is associated with increased expression of inducible nitric oxide synthase (iNOS) AFMR Meeting 2000 Carmel, CA
Vernet D et al. Spontaneous expression of inducible nitric oxide synthase (iNOS) in the hypothalamus and other brain regions of aging rats Endocrinology 1998 139: 3254–3261
Orucevic A et al. Nitric-oxide production by murine mammary adenocarcinoma cells promotes tumor-cell invasiveness Int J Cancer 1999 81: 889–896
Sasaki K et al. Nitric oxide mediates interleukin-1-induced gene expression of matrix metalloproteinases and basic fibroblast growth factor in cultured rabbit articular chondrocytes J Biochem 1998 123: 431–439
Okamoto T et al. Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: a novel mechanism for procollagenase activation involving nitric oxide Arch Biochem Biophys 1997 342: 261–274
Pelletier JP et al. Selective inhibition of inducible nitric oxide synthase in experimental osteoarthritits is associated with reduction in tissue levels of catabolic factors J Rheumatol 1999 26: 2002–2014
Racine-Samson L, Rockey DC, Bissel DM . The role of alpha1beta1 integrin in wound contraction. A quantitative analysis of liver myofibroblasts in vivo and primary culture J Biol Chem 1997 272: 30911–30917
Kurosaka H et al. Transforming growth factor-β1 promotes contraction of collagen gel by bovine corneal fibroblasts through differentiation of myofibroblasts Invest Ophtal Vis Sci 1998 39: 699–704
Faouzi S et al. Myofibroblasts are responsible for collagen synthasis in the stroma of human hepatocellular carcin-oma: an in vivo and in vitro study J Hepatol 1999 30: 275–284
Tiggelman AM et al. Transforming growth factor-beta-induced collagen synthesis by human liver myofibroblasts is inhibited by alpha 2 macroglobulin J Hepatology 1997 26: 1220–1228
Dudas J et al. Expression of decorin, transforming growth factor-beta 1, tissue inhibitor metalloproteinase 1 and 2, and type IV collagenases in chronic hepatitis Am J Clin Pathol 2001 115: 725–735
Kolb M, Margetts PJ, Sime PJ, Gauldie J . Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung Am J Physiol 2001 280: L1327–1334
He W et al. Smads mediate signaling of the TGFbeta superfamily in normal keratinocytes but are lost during skin chemical carcinogenesis Oncogene 2001 20: 471–483
Verrecchia F, Chu ML, Mauviel A . Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach J Biol Chem 2001 276: 17058–17062
Chiang PH et al. Study of the changes in collagen of the tunica albuginea in venogenic impotence and Peyronie's disease Eur Urol 1992 21: 48–51
Anafarta K et al. The significance of histopathological changes of the normal tunica albuginea in Peyronie's disease Int Urol Nephrol 1994 26: 71–77
Chiang PH et al. Study of the changes in collagen of the tunica albuginea in venogenic impotence and Peyronie's disease Eur Urol 1992 21: 48–51
Somers KD et al. Isolation and characterization of collagen in Peyronie's disease J Urol 1989 141: 629–631
Lin JS et al. Novel image analysis of corpus cavernous tissue in impotent men Urology 2000 55: 252–256
Saenz de Tejada I et al. Endothelin: localization, synthesis, activity, and receptor types in human penile corpus cavernosum Am J Physiol 1991 261: H1078–1085
Sullivan ME et al. Alterations in endothelin B receptor sites in cavernosal tissue of diabetic rabbits: potential relevance to the pathogenesis of erectile dysfunction J Urol 1997 158: 1966–1972
Iwamoto Y et al. Multiple pathways of angiotensin I conversion and their functional role in the canine penile corpus cavernosum J Pharmacol Exp Ther 2001 298: 43–48
Christ GJ, Lerner SE, Kim DC, Melman A . Endothelin-1 as a putative modulator of erectile dysfunction: I. Characteristics of contraction of isolated corporal tissue strips J Urol 1995 153: 1998–2003
Anderson MS, Shankey TV, Lubrano T, Mulhall JP . Inhibition of Peyronie's plaque fibroblast proliferation by biologic agents Int J Impot Res 2000 12: (Suppl 3) S25–S31
Gonzalez-Cadavid NF et al. Organization of the human myostatin gene and expression in healthy men and HI V-infected men with muscle wasting Proc Natl Acad Sci USA 1998 95: 14938–14943
Taylor W et al. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells Am J Physiol 2000 280: E221–E228
Ma K et al. Characterization of the 5′ upstream regulatory region of the human myostatin gene: in vitro regulation of myostatin gene transcription by dexamethasone Am J Physiol 2001 281: E1128–E1136
Artaza J et al. Nuclear localization of myostatin in C2C12 myotubes J Cell Physiol 2001 190: 170–179
Shivji R et al. Myostatin and IGF-1 and -2 expression in the muscle of rats exposed to microgravity environment of a space shuttle flight J Endocrinol 1999 167: 417–428
Kirk S et al. Myostatin regulation during skeletal muscle regeneration J Cell Physiol 2000 184: 356–363
Shapiro SD . Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease Am J Respir Crit Care Med 1994 150: S160–164
Lipshutz RJ, Fodor SPA, Gingeras TR, Lockhart DJ . High density synthetic oligonucleotide arrays Nature Genetics 1999 21: 20
Duggan D, Bittner M, Chen Y, Meltzer, Trent JM . Expression profiling using cDNA microarrays Nature Genetics 1999 21: 10
Lyer VR et al. The transcriptional program in the response of human fibroblasts to serum Science 1999 283: 83–87
Alon U et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays Proc Natl Acad Sci USA 1999 96: 6745–6751
Lee CK, Klopp RG, Weindruch R, Prolla TA . Gene expression profile of aging and its retardation by caloric restriction Science 1999 285: 1390–1395
Whitney LW et al. Analysis of gene expression in multiple sclerosis lesions using cDNA microarrays Annals Neurol 1999 46: 425–431
Lalani R, Bhasin S, Gonzalez-Cadavid NF . Expression profile of functionally related genes in skeletal muscle from rats exposed to microgravity in a spaceflight Endocrine Soc Meet 2000 San Diego, California
Taylor WE, Bhasin S, Gonzalez-Cadavid NF . Myostatin regulates expression of genes involved in skeletal muscle cell proliferation and differentiation Endocrine Soc Meet 2001 Denver, Colorado
Tanaka T et al. Expression of stress-response and cell proliferation genes in renal cell carcinoma induced by oxidative stress Am J Pathol 2000 156: 2149–2157
Hsiao LL, Stears RL, Hong RL, Gullans SR . Prospective use of DNA microarrays for evaluating renal function and disease Curr Opin Nephrol Hypertens 2000 9: 253–258
Yano N et al. Comprehensive gene expression profile of the adult human renal cortex: analysis by cDNA array hybridization kidney Int 2000 57: 1452–1459
Moch H et al. High-throughput tissue microarray analysis to evaluate genes uncovered by cDNA microarray screening in renal cell carcinoma Am J Pathol 1999 154: 981–986
Carlisle AJ et al. Development of a prostate cDNA microarray and statistical gene expression analysis package Mol Carcinog 2000 28: 12–22
Elek J, Park KH, Narayanan R . Microarray-based expression profiling in prostate tumors In Vivo 2000 14: 173–182
Xu J et al. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray Cancer Res 2000 60: 1677–1682
Saban MR et al. Time course of LPS-induced gene expression in a mouse model of genitourinary inflammation Physiol Genomics 2001 5: 147–160
Bonebaeck CAK . Antibodies in diagnostics-from immuno-assays to protein chips Immunol Today 2000 21: 379–382
Magee TR et al. Gene expression profiles in the Peyronie's disease plaque Urology 2002 59: 451–457
Chen J et al. Microarray analysis of Tbx2-directed gene expression: a possible role in osteogenesis Mol Cell Endocrinol 2001 177: 43–54
Katoh K et al. Genomic organization of the mouse OSF-1 gene DNA Cell Biol 1992 11: 735–743
Qian A et al. The gene expression profiles of Dupuytren's nodules and Peyronie's plaque suggest differential repair in related fibrotic tissues Am Soc Sex Med Meet 2001 Charleston, North Cardina
Luzzi V, Holtschlag V, Watson MA . Expression profiling of ductal carcinoma in situ by laser capture microdissection and high-density oligonucleotide arrays Am J Pathol 2001 158: 2005–2010
Wu J, Zern MA . Hepatic stellate cells: a target for the treatment of liver fibrosis J Gastroent 2000 35: 665–672
Cutroneo KR . Comparison and evaluation of gene therapy and epigenetic approaches for wound healing Wound Repair Regen 2000 8: 494–502
Tefekli A et al. The effect of decorin on the rat model of Peyronie's disease J Urol 2001 165: 200 (#828)
Kadioglu A et al. Treatment of Peyronie's disease with oral colchicine: long-term results and predictive parameters of successful outcome Int J Impot Res 2000 12: 169–175
Ahuja S et al. A pilot study demonstrating clinical benefit from intralesional interferon alpha 2B in the treatment of Peyronie's disease J Androl 1999 20: 444–448
Mignatti P, Rifkin DB, Welgus HG, Parks WC . Proteinases and tissue remodeling. In: Clark RAF, ed The molecular and cellular biology of wound repair 2nd Edition Plenum Press: New York 1996 pp 427–474
Gelbard MK, James K, Riach P, Dorey F . Collagenase versus placebo in the treatment of Peyronie's disease: a double-blind study J Urol 1993 149: 56–58
Magee TR et al. Gene therapy of erectile dysfunction in the rat with penile neuronal nitric oxide synthase (PnNOS) cDNA Biol Reprod 2002 (in press)
Robert L . Aging of the vascular wall and atherogenesis: role of the elastin-laminin receptor Atherosclerosis 1996 123: 169–179
Rehman J, Benet A, Melman A . Use of intralesional verapamil to dissolve Peyronie's disease plaque: a long-term single-blind study Urology 1998 51: 620–626
Philip S et al. Nitric oxide regulates monocyte chemotactic protein-1 Circulation 1997 96: 934–940
Sosne G et al. Thymosin beta 4 promotes corneal wound healing and modulates inflammatory mediators in vivo Exp Eye Res 2001 72: 605–608
Malinda KM et al. Thymosin beta4 accelerates wound healing J Invest Dermatol 1999 113: 364–368
Acknowledgements
The experimental studies conducted by our group were funded primarily by a grant from the Eli and Edythe L. Broad Foundation, and supported for some approaches by NIH grants R01DK-53069 and G12RR-03026.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gonzalez-Cadavid, N., Magee, T., Ferrini, M. et al. Gene expression in Peyronie's disease. Int J Impot Res 14, 361–374 (2002). https://doi.org/10.1038/sj.ijir.3900873
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ijir.3900873
Keywords
This article is cited by
-
The prevalence and topographic distribution of penile calcification in a large cohort: a retrospective cross-sectional study
International Journal of Impotence Research (2023)
-
A review of inflammation and fibrosis: implications for the pathogenesis of Peyronie’s disease
World Journal of Urology (2020)
-
Inhibition of histone deacetylase 2 mitigates profibrotic TGF-β1 responses in fibroblasts derived from Peyronie's plaque
Asian Journal of Andrology (2013)
-
Mechanisms of Disease: new insights into the cellular and molecular pathology of Peyronie's disease
Nature Clinical Practice Urology (2005)
-
Comparison of oxidative/antioxidative status of penile corpus cavernosum blood and peripheral venous blood
International Journal of Impotence Research (2005)
