
Abstract
Infection with the hepatitis B virus (HBV) leads to different disease outcomes, which can be broadly divided into three categories: acute mild infection, ‘fulminant’ and chronic hepatitis (long-term persistent form of the infection). The factors that influence the development of these different disease states are poorly understood and may include viral polymorphisms. To investigate this possibility, we analysed 116 published complete HBV genomes for which we knew disease outcome and had access to associated information on patients (age, sex and geographic origin). Our best statistical model correctly classified 72% of the cases and retained age and sex of the patient, as well as 29 candidate mutations. With the exception of one mutation in the X gene, all were located in the viral polymerase, suggesting this gene plays a critical role in clinical outcome. Our results highlight the importance of the genetics of HBV strains in the evolution of the disease and demonstrate that disease outcome can be predicted to a surprisingly large extent with a limited number of host and viral factors.
Similar content being viewed by others
Introduction
Infection with the hepatitis B virus (HBV) is a global healthcare problem with over 400 million people chronically infected (Previsani and Lavanchy, 2002). HBV can cause a variety of different outcomes. Some individuals develop an ‘acute’ hepatitis that resolves, in weeks, without secondary consequences; a small proportion suffer from a very severe hepatitis (‘fulminant’ hepatitis B) that is often fatal, while other infected individuals develop a chronic infection that causes a slowly progressive hepatitis leading in some cases to cirrhosis and liver cancer after many decades of infection. The factors that determine the outcome of infection are not yet fully understood but host and environmental factors are known to play a major role (Thursz, 2001). In particular, age at infection has a very significant impact on disease outcome and children are more likely to develop chronic infection than adults (Edmunds et al, 1993, 1996). Host genetic factors are important as studies in monozygotic twins show similarity in disease outcome but factors other than the host genotype are relevant, since some twin studies show marked differences in the course of the disease (Lin et al, 1989; Hohler et al, 2002).
Hepatitis B is a partially double stranded DNA virus with an unusual replication strategy involving an RNA intermediate that is reverse transcribed to yield DNA (Tiollais et al, 1985). This error prone replication strategy leads to a high rate of mutation within the virus, such that the virus constantly evolves both within an individual and a population (Ganem, 1996; Hannoun et al, 2000). Studies of the sequence of HBV in different populations have led to the classification of the virus into a number of different genotypes (labelled alphabetically A–H) (Kramvis and Kew, 2005) and some genotypes may be associated with different disease outcomes and different therapeutic responses (Mayerat et al, 1999; Kao et al, 2000; Hou et al, 2002; Kao, 2002; Kidd-Ljunggren et al, 2002; Fung and Lok, 2004; Jazayeri et al, 2004; Schaefer, 2005). Within individuals who are chronically infected with the virus there is evidence of on-going mutation, with substitution rates as high as 4.2 × 10−5 nucleotide substitutions per site per year (Fares and Holmes, 2002). Some of these mutations give rise to phenotypic changes – for example, the loss of hepatitis B e antigen (HBeAg) is often associated with a single point mutation in the pre-core region of the viral genome (G1896A) (Carman et al, 1989) – but the significance of the other changes during the course of an infection is unknown. It is, however, clear that the substitution rates within an individual vary and individuals with asymptomatic disease have very low rates of mutation (McIntosh et al, 1998), perhaps suggesting that the host immune response, which may be responsible for disease activity, leads to selection of preferred mutations (Lin et al, 2001, Preikschat et al, 1999). Taken together, these studies indicate that the outcome of infection with the hepatitis B virus is determined by complex interactions involving the age at infection, the viral and host genotype and the effects of the host response on viral replication (Torre and Naoumov, 1998; Gunther, 2000; Bartholomeusz and Locarnini, 2001; Kao et al, 2002; Kramvis and Kew, 2005).
Studies of the effects of viral mutations on disease phenotype are complex and are particularly difficult in chronic infections, in which multiple factors contribute to disease outcome – and where the virus mutates during an infection such that the current viral sequence may differ from the inoculating strain. To begin to address some of the many factors that determine the outcome of HBV infection, we have analysed the effects of multiple variables in patients with acute, chronic and fulminant HBV infection. Using published HBV sequences, we applied a methodology that we developed earlier to quantify the relative contribution of host factors and individual viral mutations on disease outcome (Szmaragd et al, 2006). Our results confirm previous associations between disease outcome and age and sex and, additionally, identify 29 candidate mutations within the HBV genome. A third of these mutations have been previously described and, intriguingly, all except one are found in the polymerase gene. This result may indicate that HBV outcome is linked to viral reproduction as well as the host's immune response.
Methods
The most straightforward approach to modelling clinical outcomes of an infection is to take advantage of the powerful and flexible statistical Generalised Linear Model (GLM) framework (Venables and Ripley, 1999). We consider three main outcomes acute, ‘fulminant’ and chronic hepatitis and model them with a multinomial distribution. The aim of the method is to identify the candidate viral mutations involved in the outcome of an infection. In order to decrease the number of parameters that have to be fitted, and therefore increase the statistical power of the analysis, we consider well-supported phylogenetic clades as explanatory variables (Szmaragd et al, 2006). We define each phylogenetic clade supported by a bootstrap value above 70% as a binary predictor coded for each sequence as ‘1’ for belonging to the clade and ‘0’ not belonging to it. Additional information on the host, age, sex and ethnic origin are also included in the model. We then reduce the statistical model to include only the factors that are informative at explaining clinical outcome. For all phylogenetic clades retained as predictors of clinical outcome, we identify the candidate genetic polymorphisms that support those clades (ie polymorphisms with a consistency index >75%).
Sequence alignments and phylogenetic analyses
We used the 116 complete sequences of human HBV available from Genbank in March 2005 for which we could obtain information on the outcome of the infection, as well as age, sex and geographic origin of the patient (Supplementary Table S1). Amino–acid sequences were aligned using MEGA3 (Kumar et al, 2004), considering each gene separately to respect the reading frames. The parameter values for the alignments were identical to the one used by Szmaragd et al (2006). In addition to all possible amino-acids, we considered stop codons and gaps as phylogenetically informative states. The phylogenies were constructed separately for each gene using PAUP* (Swofford, 2002) with maximum parsimony criterion and assessed by 2000 bootstrap replicates. The consensus trees based on a 50% majority-rules were drawn and annotated with TreeDyn (Chevenet, 2006). The phylogenetics algorithms – heuristic search with 10 replicates of random-sequence-addition for each tree-bisection-reconnections – are identical to the ones described in Szmaragd et al (2006). We used maximum parsimony as this is the only phylogenetic method that allows for direct characterisation of the association between individual mutations and clades within a tree. In order to assess the robustness of our phylogenetic reconstructions, we also analysed the same data set with neighbour joining with an underlying JTT matrix, and gamma rate variation among sites. Both methods recovered the same clades with an essentially identical general topology (results not shown).
HBV is composed of four overlapping reading frames (ORFs) commonly named polymerase, precore/core (pc/core), envelope (preS1/S2/S genes) and the X gene. As each of those genes might influence the outcome of the infection, we tested whether their phylogenetic information content was consistent, using an Incongruence Length Difference (ILD) Test (Cunningham, 1997) in PAUP*. The four genes were shown to be incongruent (P<0.0001), implying that a separate phylogeny has to be constructed for each individual gene. It has been recently proposed that recombination might be extensive in the HBV virus (Simmonds and Midgley, 2005), and there is no reason to believe that all recombination breakpoints should lie at the boundaries between genes. However, recombination is expected to be a major issue when estimating underlying parameters such as divergence times. In this work, our application of phylogenetics is far less ambitious as we only aim at defining related clusters of strains. In such a context, recombination is not expected to be a major problem. Similarly, we did not split genes into parts overlapping with other ORFs versus nonoverlapping regions. Indeed, we previously modelled genetic diversity, substitution rate and other parameters such as codon bias for the entire HBV genome. While one would expect sites in overlapping regions to be more constrained, whether a site was situated within overlapping ORFs seems to have very little effect. Somewhat unexpectedly, there was even a tendency for overlapping sites to be characterised by a slightly higher substitution rates (Szmaragd and Balloux unpublished).
Statistical analyses
We fitted multinomial generalised linear models using the package nnet (Venables and Ripley, 1999) within the R environment (v2.1.0, R core development Team, 2004). The clinical outcome of the infection was classified into three categories: ‘fulminant’, chronic and acute hepatitis. In multinomial models, one category is arbitrarily chosen as a reference for the estimation of the coefficients of the others. We always considered acute as the baseline category, since acute hepatitis is the milder outcome of the infection. The host-related factors considered were age at sampling, sex and geographic origin (which was divided into Asia, Caucasian, Americas). Asia was chosen as a reference level for the geography variable, as it was the group with most individuals. We only considered the interaction between age and sex, as the data set was too small to investigate possible interactions involving geography.
In a first step, we built a minimal model of clinical outcome solely based on host-related factors (age, sex and geographic origin of the infected patient). We then considered the influence of the phylogenetic clades for each gene separately. We included all clades with a bootstrap support of at least 70%. Finally, a genome-wide consensus model was built. In this model, we started by including age, sex, geography and the age-sex interaction, and then added all clades found to be significant in the individual gene models, rather than inferring a phylogenetic tree for the full-genome).
Optimal models for all GLMs, defined by the lowest Akaike Information Criterion (AIC), were selected using a backward stepwise approach, starting with a fully saturated model and sequentially dropping factors that were not informative at explaining clinical outcome. To assess the predictive power of the optimal GLM models, we performed a ‘remove one’ cross-validation procedure based on a resampling method. Each observation is removed in turn, and the parameters of the minimal model are reset using the remaining observations. These new parameters are then used to predict the outcome for removed observations. This provides us with the number of cases correctly predicted by the model within each category. We express the proportion of correctly predicted cases by a particular model as an average over the three classes. The proportion of well-predicted cases (Pcorr) is given as:

where nA, nC and nF represent the total number of observations within the three categories acute, chronic and fulminant and nA′, nC′ and nF′ the number of correctly predicted cases by the cross validation process for each category. This weighting was chosen to compensate for the difference in size of the three outcome categories. The same weight is, therefore, attributed to each outcome.
Results
Host factors
The best model including only host factors retained age and geography. Sex did not have a significant effect on the outcome. The absence of a difference between males and females in terms of infection outcome can also be confirmed with a simple χ2 test that ignores other factors (P=0.56). This host-related factor-only model correctly predicts 41.9% of the cases (Figure 1a; Table 1). The model cannot predict any acute case; instead, almost all acute cases are predicted as chronic (13 chronic and one fulminant). In terms of statistical significance, this model outperforms a null model (clinical outcome ∼1) with a P-value of 5.62 × 10−6. Adding a factor for the genotype classification (A–H) into this model increased the predictive power to 47%.

Plots of predicted versus observed number of cases of disease outcomes for the different models. The number of observed cases is given by the height of the columns; the number of predicted cases by the color. The predicted numbers of cases were obtained through a cross-validation procedure as described in the material and methods. (a) Host-factors-only model, (b) polymerase, (c) preS1/S2/S gene, (d) pc/core gene, (e) X gene and (f) complete genome model.
Individual genes
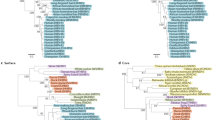
In a second step, models were fitted for each of the four genes individually (Table 1). Gene-specific phylogenetic trees are given in Figure 2. Age was retained as a key factor in all four models. Interestingly, patient ethnicity (geography), which was associated to clinical outcome in the model based on host-related factors-only, was not informative in any of the four models including viral genetic information. Thus, the viral phylogenetic classification captures more information on geographic variation in clinical outcome than the patient's ethnic classification itself. Sex and the interaction between sex and age were also retained in all models except for pc/core, for which only age was retained. All models performed better than the host-related factor-only model. The gain was most significant for the polymerase gene. Out of the 30 clades with bootstrap support over 70%, 13 were statistically associated with clinical outcome. Including those, 13 classes allows us to predict 70% of the outcomes correctly (Figure 1b; Table 1). The second best model involves the X gene. Using the four clades retained by the model allows us to predict 55.8% (Figure 1e; Table 1). Accounting for information from the preS1/S2/S or pc/core regions leads only to marginal gains in predictability of disease outcome, as both models correctly classify <50% cases (Figure 1c and d; Table 1). The poor performance of the preS1/S2/S model is surprising as this region has been associated with a functional protein that may modify the cellular response (Kekule et al, 1990). This result cannot be explained by a poor resolution of the phylogeny for this gene, since the phylogenetic tree harbours 21 clades with high bootstrap support (Figure 2b), seven of them being retained by the model.

Phylogenetic trees for the four open reading frames of hepatitis B. Terminal branches of the trees are drawn in different colors depending on clinical outcome (blue: chronic; green: acute and red: ‘fulminant’. Red circles and squares highlight clades with bootstrap support above 70 and 90% respectively. The genotypes are indicated by symbols at the tip of each branch in the tree, genotype A is represented by •, genotype B by ▪, genotype C by ○, genotype D by □ and genotype G by  .
.
Candidate mutations
An exhaustive list of all mutations that were found to be statistically associated to disease outcome is provided in Table 2. Interestingly, none of the mutations retained by our models included insertions or deletions (indels). Most candidate mutations are found in the polymerase, for which 33 candidate polymorphisms have been retained on a total of 280 possible parsimony informative sites (ie sites at which there are at least two different amino-acids, with the rarest found at least in two distinct sequences) (11.8%). Among those candidates, 13 have been previously described, mainly in drug-resistance studies (Hasegawa et al, 1994; Gunther et al, 1998; Gerner et al, 1999; Koseki et al, 1999; Hannoun et al, 2000). As a result of the overlapping reading-frames, 15 of the 33 candidate mutations in polymerase also correspond to changes in other genes: one within the X gene; 13 within the preS1/S2/S ORF (6 of them already described in the literature); and one within the pc/core ORF.
In all 18 candidate mutations were retained in the preS1/S2/S model out of a total of 152 parsimony informative sites (11.8%). Among these candidates, 10 have been previously described as epitopes (Kuroki et al, 1990; Meisel et al, 1994; Gripon et al, 2005) or as characteristic of chronic or fulminant hepatitis (Gunther et al, 1998; Koseki et al, 1999; Weinberger et al, 2000; Torresi, 2002). Only a single candidate mutation was found in the pc/core gene out of 53 parsimony informative sites. This single pc/core candidate has been previously described as affecting the translation of the HBe antigen in chronic hepatitis (Dumpis et al, 2001). For the X gene, two candidates were retained out of a total of 55 sites (3.6%). One of those corresponds to three different substitutions occurring at the same amino acid. Mutations at this position have been previously linked to α-interferon therapy success (Gunther et al, 1998).
Complete genome
For the genome-wide model, we started with the 26 clades from all four genes and the host factors (age, sex the interaction between age and sex, and geographic origin). Dropping terms successively allowed us to identify a subset of clades that are key predictors of clinical outcome. The main characteristics of this model are presented in the last row of Table 1. The best genome-wide model retained all three host factors (age, sex and their interaction) and 12 clades from the polymerase and one from the X gene. The model is highly significant (P=2.5 × 10−17) and underlines the importance of the polymerase gene, which contributes nearly exclusively to the model. This model correctly predicts 72% of the outcomes, a marginally better figure than the polymerase-based model. Adding to the model a factor representing the classification into genotypes (A–H) does not increase the predictive power of the model. The polymorphisms corresponding to the complete genome model are indicated in bold in Table 2. The clades retained are characterised by 29 candidate polymorphisms (one amino acid in the X gene and 28 in the polymerase gene), corresponding to 5.3% of a total 540 parsimonious sites. All these 28 polymerase mutations were already present in the polymerase-based model. Nine of these mutations have been previously described in the literature (Table 2).
Discussion
In this paper, we aimed at characterising both host demographic factors and HBV candidate mutations associated to disease outcome. Our best models were remarkably successful at predicting outcome, with over 70% of cases assigned correctly. We noted that age and sex are involved in the outcome of HBV infection, and we identified 29 candidate mutations in the HBV genome associated to a particular clinical outcome. All those mutations but one were found in the polymerase gene. Only a third of these mutations have been previously reported. The results presented here are a direct extension of the work presented in Szmaragd et al (2006), where we did apply the same methodology to sequences of the polymerase gene of 65 HBV strains. Here, we nearly doubled the number of strains and extend the analysis to the entire genome. The increased sample size allows us to characterise a larger number of candidate mutations and also provides far better support to the statistical models.
It is well established that infection in childhood predisposes to chronic infection (Edmunds et al, 1993, 1996; Sohn et al, 2005). However, in our study, age was positively related to chronicity. This apparent contradiction stems from us considering the age of the patients at which sampling took place and not the age at which infection occurred (which is often unknown for chronic patients). There is no such problem for the association between age and the risk of ‘fulminant’ hepatitis B, as this form of severe, often fatal hepatitis, develops within a few weeks of infection. Our analysis indicates that the relative risk of a fulminant infection decreases with age. This is contrary to the situation with other hepatotropic viruses, such as hepatitis A, where the risk of fulminant hepatitis increases with age (Howard, 2002). These data are compatible with current models suggesting that hepatitis A is directly cytopathic (Cromeans et al, 1989) and the severity of the infection may be reduced by a vigorous host immune response whereas hepatitis B is believed to be non-cytopathic and liver damage is caused by immune-mediated cytopathic effects (Bartholomeusz and Locarnini, 2001). Our model also predicts that male patients are at higher risk of developing a chronic infection than females, whereas the opposite tendency is observed for the risk of fulminant outcomes. However, those trends are not independent of age as documented by the significant interaction between age and sex.
Whenever we included information on viral genetics, the ethnicity of the patient disappeared from the models. While this does not necessarily imply that geographic variation in the host (both genetic and cultural) is irrelevant to clinical outcome, it does suggest that genetic variation in HBV genotypes is far more important than the rough classification, we have considered for human hosts. HBV is itself geographically highly clustered with specific strains (genotypes) being characterised by localised geographic distributions (Kidd-Ljunggren et al, 2002; Kramvis and Kew, 2005). Thus, there is a level of redundancy in the information provided by spatial structuring of host and pathogen variation. In the future, a finer grain classification of host's genetic variability (Manica et al, 2005) would provide a fairer test to what extent the genetic makeup of human populations plays a role in determining clinical outcome. We also observe that adding a factor for the HBV genotype classification (A–H) to the model does not lead to any improvement. This suggests that the 29 candidate mutations we detect capture the effect of HBV genetics on disease outcome. The publicly available data we use is obviously not a random sample. Severe cases are likely to be highly overrepresented. This should not be a problem, as we do not make inferences on probabilities of specific disease outcome, but on factors that do correlate with them.
A striking feature of the full genome model is that it almost exclusively retained clades from the polymerase (with the exception of only one X gene clade). While this does not imply that mutations in preS1/S2/S and pc/core have no influence on the clinical outcome, their contribution is weak compared to polymerase gene. This conclusion is supported by the relatively poor predictive power of the models specifically considering the preS1/S2/S and pc/core genes. Our methodology might have marginally more power at detecting candidate mutations in longer genes, as its power is expected to increase for well-resolved phylogenies. The polymerase is indeed longer than the other three genes considered as it encompasses two thirds of the HBV genome. However, this source of bias should be minimal for the preS1/S2/S gene, which is characterised by a well-resolved phylogeny including 21 clades with a bootstrap support over 70%. One possible explanation for the increase in ‘outcome associated’ polymorphisms within the polymerase gene is that patients with chronic HBV receiving therapy were more likely to be sequenced than those who were not receiving treatment and that such patients are more likely to contain mutations within the polymerase, perhaps selected as conferring a survival advantage in the presence of a therapeutic agent. Such a bias in subject selection may have led to a high frequency of patients on therapy (ie those with chronic infection) being entered onto the publicly available databases. We believe that this explanation is unlikely as studies of drug resistance almost invariably focus exclusively on the enzymatic regions within the polymerase gene and we included only cases where sequence data for the entire virus was available. Database annotations indicated that only 10 patients were receiving therapy at the time of sequencing. Besides, the majority of mutations within the polymerase gene were not present in enzymatically active domains that have been implicated in resistance to antiviral agents. Thus, we believe that nonrandom selection of patients receiving therapy cannot explain our findings, although we cannot exclude the possibility that drug induced mutations made a minor contribution to our findings.
The polymerase plays a key role in the biological cycle of the virus, as it is responsible for the replication process (Tiollais et al, 1985). Thus, any mutation within this ORF can have a direct effect on replication efficiency, impacting the fitness of the virus (Torresi, 2002). Such a direct effect on virus replication capacity might be invoked for several of the candidate mutations we characterize in the HBV polymerase. However, nearly half of the polymerase mutations are located in the spacer domain, a region that is not generally believed to be important in virus replication. This observation could be due to three nonexclusive factors. Firstly, those mutations might be false positives, ‘hitch hiking’ in association with genuine mutations that affect disease outcome. Secondly, they might reflect important mutations in the envelope (Torresi et al, 2002) as the spacer domain overlaps the envelope genes and also key promoter regions. In all, 13 of the mutations within the spacer correspond to nonsynonymous changes within the preS1/S2/S ORF, but only three of those were retained by the preS1/S2/S model. A number of the mutations in the spacer, while they do not directly affect the ORF of the envelope genes, may affect the promoters and therefore the levels of the gene products. The balance between the three viral proteins is very important for viral secretion and has been shown to be affected by mutations in the promoters (Xu and Yen, 1996). Functional analysis will be required to determine whether some of the mutations described here affect the promoter activity and virion secretion. Thirdly, the spacer domain might simply be more important for polymerase efficacy than generally accepted.
Previous studies on the mutations within HBV that modify disease outcome have noted the strong correlation between mutations in the precore region of HBV (in particular G1896A) and the presence of HBeAg negative disease (Carman et al, 1997; Blackberg and Kidd-Ljunggren, 2000). In our study, we focused on the differences between acute, chronic and ‘fulminant’ infection and we found no association with mutations known to modify HBeAg formation and any of these disease states. This suggests that the presence or absence of HBeAg may not impact upon the initial outcome of exposure to HBV although it is clear from previous studies that it does have a major impact on the further development of chronic infection (Lindh et al, 1996; Tsubota et al, 1998; Hou et al, 2002). The fact that we did not detect G1896A as a candidate mutation should not be interpreted as a failure of our methodology. Indeed a simple χ2 test on the frequency of A and G at position 1896 in the 116 genomes we analysed shows no significant association with disease outcome.
Understanding the factors influencing the clinical outcome of infectious diseases is crucial for early diagnosis and optimised treatment. Despite widespread recognition of the importance of the genetic composition of infecting strains and host factors such as age or sex, most efforts so far have focused narrowly on characterising disease and susceptibility genes in humans. We feel there is great need to develop methodologies that take into account both factors from the host and the pathogen. Our results suggest that the genetic composition of the infecting HBV strains is a major determinant of clinical outcome. We could further characterise a list of 29 mutations statistically associated to disease outcome, all but one in the polymerase gene. Many of these candidate mutations seem unexpected given our current knowledge of the molecular genetics of HBV. Thus, it remains to be seen whether functional analyses will confirm their role in modifying the course of infection.
References
Bartholomeusz A, Locarnini S (2001). Hepatitis B virus mutants and fulminant hepatitis B: Fitness plus phenotype. Hepatology 34: 432–435.
Bartholomeusz A, Locarnini S, Ayres A, Thompson G, Sozzi V, Angus P et al (2004). Molecular modelling of hepatitis B virus polymerase and adefovir resistance identifies three clusters of mutations. Hepatology 40: 246A.
Blackberg J, Kidd-Ljunggren K (2000). Genotypic differences in the hepatitis B virus core promoter and precore sequences during seroconversion from HBeAg to Anti- HBe. J Med Virol 60: 107–112.
Carman W (1997). The clinical significance of surface antigen variants of hepatitis B virus. J Viral Hepatitis 4: 11–20.
Carman W, Boner W, Fattovich G, Colman K, Dornan E, Thursz M et al (1997). Hepatitis B virus core protein mutations are concentrated in B cell epitopes in progressive disease and in T helper cell epitopes during clinical remission. J Infect Dis 175: 1093–1100.
Carman W, Jacyna M, Hadziyannis S, Karayiannis P, McGarvey M, Makris A et al (1989). Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 2: 588–589.
Chayama K, Suzuki Y, Kobayashi M, Kobayashi M, Tsubota A, Hashimoto M et al (1998). Emergence and takeover of YMDD motif mutant hepatitis B virus during long-term lamivudine therapy and re-takeover by wild type after cessation of therapy. Hepatology 27: 1711–1716.
Chevenet F (2006). TreeDyn: towards dynamic graphics & annotations for trees analyses V194.3 [http://www.treedyn.org].
Choi B, Parker G, Rho H (1999). Interaction of hepatitis B viral X protein and CCAAT/ enhancer-binding protein alpha synergistically activates the hepatitis B viral enhancer II/pregenomic promoter. J Biol Chem 29: 2858–2865.
Cromeans T, Fields H, Sobsey M (1989). Replication kinetics and cytopathic effect of hepatitis A virus. J Gen Virol 70: 2051–2062.
Cunningham C (1997). Can three incongruence tests predict when data should be combined? Mol Biol Evol 14: 733–740.
Dumpis U, Mendy M, Hill A, Thursz M, Hall A, Whittle H et al (2001). Prevalence of HBV core promoter/precore/core mutations in Gambian chronic carriers. J Med Virol 65: 664–670.
Edmunds W, Medley G, Nokes D, Hall A, Whittle H (1993). The influence of age on the development of the hepatitis-B carrier state. Proc R Soc Lond Ser B-Biol Sci 253: 197–201.
Edmunds W, Medley G, Nokes D, OCallaghan C, Whittle H, Hall A (1996). Epidemiological Patterns of hepatitis B virus (HBV) in highly endemic areas. Epidemiol Infect 117: 313–325.
Fares M, Holmes E (2002). A revised evolutionary history of hepatitis B virus (HBV). J Mol Evol 54: 807–814.
Ferrari C, Bertoletti A, Penna A, Cavalli A, Valli A, Missale G et al (1991). Identification of immunodominant T cell epitopes of the hepatitis B virus nucleocapsid antigen. J Clin Invest 88: 214–222.
Ferrari C, Cavalli A, Penna A, Valli A, Bertoletti A, Pedretti G et al (1992). Fine specificity of the human T-cell response to the hepatitis B virus preS1 antigen. Gastroenterology 103: 255–263.
Fung S, Lok A (2004). Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 40: 790–792.
Ganem D (1996). Hepadnaviridae and their replication. In: Fields B, Knipe D and Howley P (eds) Virology, 3rd edn. Lippincott-Raven Publishers: New York. pp 2703–2737.
Gerner P, Lausch E, Friedt M, Tratzmuller R, Spangenberg C, Wirth S (1999). Hepatitis B virus core promoter mutations in children with multiple anti-HBe/HBeAg reactivations result in enhanced promoter activity. J Med Virol 59: 415–423.
Gripon P, Cannie I, Urban S (2005). Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J Virol 79: 1613–1622.
Gunther S (2000). Naturally occurring mutations of hepatitis B virus and outcome of chronic infection: Is there an association? Commentary Eur J Clin Invest 30: 751–753.
Gunther S, Paulij W, Meisel H, Will H (1998). Analysis of hepatitis B virus populations in an interferon-alpha-treated patient reveals predominant mutations in the C-gene and changing e-antigenicity. Virology 244: 146–160.
Hannoun C, Horal P, Lindh M (2000). Long-term mutation rates in the hepatitis B virus genome. J Gen Virol 81: 75–83.
Hasegawa K, Huang J, Rogers S, Blum H, Liang T (1994). Enhanced replication of a hepatitis-B virus mutant associated with epidemic of fulminant hepatitis. J Virol 68: 1651–1659.
Hohler T, Reuss E, Evers N, Dietrich E, Rittner C, Freitag C et al (2002). Differential genetic determination of immune responsiveness to hepatitis B surface antigen and to hepatitis A virus: a vaccination study in twins. Lancet 360: 991–995.
Hou J, Lin Y, Waters J, Wang Z, Min J, Liao H et al (2002). Detection and significance of a G1862T variant of hepatitis B virus in Chinese patients with fulminant hepatitis. J Gen Virol 83: 2291–2298.
Howard C (2002). Hepatitis viruses: a Pandora's box? J Gastroenterol Hepatol 17: S464–S467.
Jazayeri M, Basuni A, Sran N, Gish R, Cooksley G, Locarnini S et al (2004). HBV core sequence: definition of genotype-specific variability and correlation with geographical origin. J Viral Hepatitis 11: 488–501.
Kao J (2002). Hepatitis B viral genotypes: clinical relevance and molecular characteristics. J Gastroenterol Hepatol 17: 643–650.
Kao J, Chen P, Lai M, Chen D (2000). Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118: 554–559.
Kao J, Chen P, Lai M, Chen D (2002). Genotypes and clinical phenotypes of hepatitis B virus in patients with chronic hepatitis B virus infection. J Clin Microbiol 40: 1207–1209.
Kekule A, Lauer U, Meyer M, Casselmann W, Hofschneider P, Koshy R (1990). The preS2/s region of integrated hepatitis-B virus DNA encodes a transcriptional transactivator. Nature 343: 457–461.
Kidd-Ljunggren K, Miyakawa Y, Kidd A (2002). Genetic variability in hepatitis B viruses. J Gen Virol 83: 1267–1280.
Koseki T, Hongo S, Muraki Y, Sugawara K, Matsuzaki Y, Nakamura K (1999). Sequence analysis of the entire genome of hepatitis B virus from a patient with fulminant hepatitis. Yamagata Med J 17: 27–40.
Kramvis A, Kew M (2005). Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J Viral Hepatitis 12: 456–464.
Kumar S, Tamura K, Nei M (2004). Mega3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings Bioinformat 5: 159–163.
Kuroki K, Floreani M, Mimms L, Ganem D (1990). Epitope mapping of the Pres1 domain of the hepatitis-B virus large surface protein. Virology 176: 620–624.
Lin TM, Chen CJ, Wu MM, Yang CS, Chen JS, Lin CC et al (1989). Hepatitis-B virus markers in Chinese twins. Anticancer Res 9: 737–741.
Lin X, Qian G, Lu P, Wu L, Wen Y (2001). Full length genomic analysis of hepatitis B virus isolates in a patient progressing from hepatitis to hepatocellular carcinoma. J Med Virol 64: 299–304.
Lindh M, Horal P, Dhillon A, Furuta Y, Norkrans G (1996). Hepatitis B virus carriers without precore mutations in hepatitis B e antigen-negative stage show more severe liver damage. Hepatology 24: 494–501.
Manica A, Prugnolle F, Balloux F (2005). Geography is a better determinant of human genetic differentiation than ethnicity. Hum Genet 118: 366–371.
Mayerat C, Mantegani A, Frei P (1999). Does hepatitis B virus (HBV) genotype influence the clinical outcome of HBV infection? J Viral Hepatitis 6: 299–304.
McIntosh E, Givney R, Zhang S, Courouce A, Burgess M, Cossart Y (1998). Molecular epidemiology and variation of hepatitis B in recent immigrant families to Australia. J Med Virol 56: 10–17.
Meisel H, Sominskaya I, Pumpens P, Pushko P, Borisova G, Deepen R et al (1994). Fine mapping and functional characterization of 2 immunodominant regions from the PreS2 sequence of hepatitis B virus. 37: 330–339.
Oess S, Hildt E (2000). Novel cell permeable motif derived from the PreS2-domain of hepatitis-B virus surface antigens. Gene Therapy 7: 750–758.
Preikschat P, Meisel H, Will H, Gunther S (1999). Hepatitis B virus genomes from long-term immunosuppressed virus carriers are modified by specific mutations in several regions. J Gen Virol 80: 2685–2691.
Previsani N, Lavanchy D (2002). Hepatitis B: World Health Organization, Department of Communicable Diseases Surveillance and Response [http://www.who.int/csr/en/].
R Core Development Team (2004). A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna.
Raney A, Le H, McLachlan A (1992). Regulation of transcription from the hepatitis B virus major surface antigen promoter by the Sp1 transcription factor. J Virol 66: 6912–6921.
Rehermann B, Fowler P, Sidney J, Person J, Redeker A, Brown M et al (1995). The cytotoxic T lymphocyte response to multiple hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp Med 181: 1047–1058.
Schaefer S (2005). Hepatitis B virus: significance of genotypes. J Viral Hepatitis 12: 111–124.
Shaul Y, Ben-Levy R, De-Medina T (1986). High affinity binding site for nuclear factor I next to the hepatitis B virus S gene promoter. Embo J 5: 1967–1971.
Simmonds P, Midgley S (2005). Recombination in the genesis and evolution of hepatitis B virus genotypes J. Virol 79: 15467–15476.
Sohn J, Lee C, Lee J, Lee K, Son C, Lee J et al (2005). Mutation analysis of hepatitis B virus promoters in chronically infected children. Arch Virol Suppl 150: 1639–1651.
Swofford D (2002). PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) 4.0b10 edn. Sinauer Associates: Sunderland, MA.
Szmaragd C, Nichols R, Balloux F (2006). A novel approach to characterise pathogen candidate mutations involved in clinical outcome. Infect Genet Evol 6: 38–45.
Thursz M (2001). Genetic susceptibility in chronic viral hepatitis. Antiviral Res 52: 113–116.
Tiollais P, Pourcel C, Dejean A (1985). The Hepatitis-B virus. Nature 317: 489–495.
Torre F, Naoumov N (1998). Clinical implications of mutations in the hepatitis B virus genome. Eur J Clin Invest 28: 604–614.
Torresi J (2002). The virological and clinical significance of mutations in the overlapping envelope and polymerase genes of hepatitis B virus. J Clin Virol 25: 97–106.
Torresi J, Earnest-Silveira L, Deliyannis G, Edgtton K, Zhuang H, Locarnini S et al (2002). Reduced antigenicity of the hepatitis B virus HBsAg protein arising as a consequence of sequence changes in the overlapping polymerase gene that are selected by lamivudine therapy. Virology 293: 305–313.
Tsubota A, Kumada H, Takaki K, Chayama K, Kobayashi M, Kobayashi M et al (1998). Deletions in the hepatitis B virus core gene may influence the clinical outcome in hepatitis B e antigen positive asymptomatic healthy carriers. J Med Virol 56: 287–293.
Venables W, Ripley B (1999). Modern Applied Statistics with S. Springer: New York.
Weinberger K, Bauer T, Bohm S, Jilg W (2000). High genetic variability of the group-specific a-determinant of hepatitis B virus surface antigen (HBsAg) and the corresponding fragment of the viral polymerase in chronic virus carriers lacking detectable HBsAg in serum. J Gen Virol 81: 1165–1174.
Xu Z, Yen T (1996). Intracellular retention of surface protein by a hepatitis B virus mutant that releases virion particles. J Virol 70: 133–140.
Acknowledgements
We are indebted to William Bower, Richard Garfein and Hideaki Kato, who agreed to share unpublished information with us. We also thank François Chevenet for his help with the program Treedyn, and two anonymous reviewers for helpful comments. CS is supported by the Isaac Newton Trust, the Cambridge European Trust, and the British Federation of Women Graduates.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Szmaragd, C., Foster, G., Manica, A. et al. Genome-wide characterisation of Hepatitis B mutations involved in clinical outcome. Heredity 97, 389–397 (2006). https://doi.org/10.1038/sj.hdy.6800882
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800882
