
Abstract
Homologous markers on the sex-specific regions of the X- and Y-chromosomes are differentially inherited through males and females, and have similar molecular characteristics. They may therefore be useful as a complement to the comparison of mtDNA and Y-chromosomal haplotypes for estimating sex-specific processes shaping human population structure. To test this idea, we analyzed XY-homologous microsatellite diversity in 33 human populations from Africa, Asia and Europe. Interpopulation comparisons suggest that the generally discordant pattern of genetic variation observed for X- and Y-linked markers could be an outcome of sex-specific migration processes (mfemales/mmales∼3) or sex-specific demographic processes (Nfemales/Nmales∼11) or a combination of both. However, intrapopulation diversity estimated by the X/Y ratio Watterson estimator (θH(Y)/θH(X)) suggests that the scenarios required to explain the global genetic variation of XY-homologous markers are many and complex, and that the sex-specific processes (effective population size and migration rate) shaping human population structures are likely to be specific to each population under study. XY-homologous markers provide an insight into the genuine complexity of sex-specific processes, and their further exploitation in human population studies seems worthwhile.
Similar content being viewed by others

Introduction
Mitochondrial and Y-chromosomal markers show large differences in a way that genetic variation is partitioned within and between populations – intrapopulation diversity is usually lower for the Y-chromosome (inherited uniparentally through males) than for mtDNA (inherited uniparentally through females) (eg Jorde et al, 2000; Kayser et al, 2003). These differences could be due to sex-specificity in rate and scale of migration (including the prevalence of patrilocal marriage practices) and in effective population size (eg Bamshad et al, 1998; Seielstad et al, 1998; Perez-Lezaun et al, 1999; Oota et al, 2001; Kayser et al, 2003); consequently, comparisons between mtDNA and the Y-chromosome have been widely used to characterize sex-specific patterns. However, the differences could also reflect selective pressures acting differently in the two genomic components as well as their great disparity in molecular characteristics (such as size, copy number, cellular localization, coding proportion and mutation rate) (Cummins, 2001). The discordant conclusions about male and female migration rates and effective population sizes reached by studies using similar mtDNA markers but different Y-chromosomal markers provide an illustration of this problem (Wilder et al, 2004; Wood et al, 2005).
To test to what extent sex-specific processes shape the genetic structure of human populations, it would be desirable to study the variability of markers that are comparable to each other in molecular terms, and located in similar genomic contexts. In addition, to avoid bias arising from differential selection, it would be important to possess information about the selective pressures acting on these markers.
In the present study, we consider an attractive candidate for such a model, markers on the X- and Y-chromosomes, which share homology through an autosomal common origin. This homology ensures they share similar molecular characteristics. The differentiation of the sex chromosomes started 320 MA ago, presumably with a local suppression of recombination between the proto-X and proto-Y (Lahn and Page, 1999). A series of up to five successive inversions were responsible for the complete suppression of recombination between the X- and Y-chromosomes, with the exception of the pseudoautosomal region(s) (Ross et al, 2005). Although sequence divergence resulted from recombination suppression, some non-pseudoautosomal regions have conserved a very high level of DNA similarity (Ross et al, 2005). Exploiting the ancestral homology of the human sex chromosomes and their modes of inheritance (2/3 of X-chromosomes are inherited from females and 1/3 from males), we propose to use XY-homologous markers as a complementary tool to mtDNA and Y-chromosome to estimate the sex-specific processes shaping the genetic structure of human populations. We focus on the Xp22.3 and Yq11 homologous region in which dinucleotide microsatellite markers have previously been characterized (Malaspina et al, 1997) and studied (Scozzari et al, 1997). These markers are attractive because they can be specifically amplified from each chromosome, and therefore provide unambiguous information about both X- and Y-chromosome diversity. Their similar genomic contexts have been already described (Balaresque et al, 2003).
In this study, we analyze XY-linked microsatellite markers in 33 populations from Africa, Europe and Asia. We carry out an extensive intra- and interpopulation genetic differentiation analysis and evaluate alternative sex-specific processes to explain the observed patterns, asking if these processes differ according to the continental area under study. Our purpose is not to draw conclusions about the precise processes involved in the history of each population, but to assess the potential of XY-homologous markers, and to ask if the development of further marker systems from the extensive available sequence data is worthwhile.
Materials and methods
Samples
A total data set of 2549 X-chromosomes and 1413 Y-chromosomes from Africa, Europe and Asia was analyzed, representing 33 populations located in eight major geographic regions in Europe, Africa and Asia (Figure 1). Most of these data were extracted from Scozzari et al (1997) and Balaresque et al (2004). To complete the data published in the latter paper, we genotyped 386 Y-chromosomes from Ivory Coast (Akan and Yacouba), Ethiopia (Amhara and Oromo), Algeria (Mozabites), Morocco (Berbers), Corsica and the Orkney Islands (Orcadians). These males have been already typed for the X-linked microsatellite.

Map showing the approximate location of 33 populations studied. Data extracted from Scozzari et al (1997) are represented by black circles; white circles refer to the data extracted from Balaresque et al (2004) and completed in the present study. The circles indicate the geographical area considered.
PCR amplification
The DYS413a,b microsatellite amplifications followed the protocols of Scozzari et al (1997). The DYS413 polymorphic system corresponds to two coamplified Y-specific loci each containing a (CA)n microsatellite. Alleles were treated according to Mathias et al (1994): the larger PCR fragment generated was assigned to the allelic classes DYS413a and the smaller to DYS413b. Whenever a single band was observed, the presence of two fragments of the same size was assumed. PCR primers were fluorescently labelled and the PCR products were run in a standard 6% denaturing polyacrylamide gel using an ABI 373A automated sequencer (Applied Bio-systems). GeneScan and Genotyper software packages (Applied Bio-systems) were used to size the amplified alleles.
Information on genomic context for the XY-homologous microsatellites and calculation of the Y/X mutation rate
The (CAIII) microsatellites have been mapped to Xp22.3 and Yq11, are located in one of the homologous regions existing between X- and Y-chromosomes outside the pseudoautosomal regions I and II, and are shown in Figure 2a. This representation is based on recent information about the molecular structure of the Y-chromosome (Skaletsky et al, 2003) and the X-chromosome (Ross et al, 2005). By an in silico search, we confirmed the location of the DXS8175 microsatellite in the homology region 9 (Ross et al, 2005) close to the VCX-B1 (VCX10r). On the Y-chromosome, the DYS413a,b microsatellites are located within palindrome P8, which shows arm to arm identity of 99.997% (Skaletsky et al, 2003). Figure 2b shows each microsatellite studied within XY-homologous region and the nearby VCXY genes.

(a) The microsatellites and their surrounding sequences (∼3 kb) are represented by the black box. The homology region is represented by the two gray boxes on X- and Y-chromosomes. The detailed structure of the homology region on each chromosome is reported: on the Y-chromosome, we represented the two arm of the palindrome P8 (according to Skaletsky et al, 2003), and on the X-chromosome, the different subpart of homologous regions (according to Ross et al, 2005). (b) The molecular structure of the CAIII microsatellites and their surrounding sequences (containing VCXY genes) are represented.
This high degree of sequence similarity between the homologous flanking sequences of the X- and Y-linked microsatellites allows us to calculate the ratio of mutation rates between the sequences. This was carried out by counting the number of Y/X nucleotide substitutions in the flanking sequences in different primate species (Balaresque et al, 2003), and converting this to a mean substitution rate by considering the species divergence times, taken from Chen and Li (2001). Using comparisons between human and chimpanzee, human and gorilla and human and orangutan, the mean nucleotide substitution rates were 0.03 substitutions/site/MYA for DXS8175 and 0.20 substitutions/site/MYA for DYS413a,b. The Y/X ratio is then 0.20/0.03=6.6. This value is higher than those reported in the literature (see, for a review, Li et al, 2002), and this point will be addressed in the Discussion.
Measures of genetic diversity
Estimates of intrapopulation diversity
Genetic diversity (h) was estimated as h=1−∑pi2, where pi is the estimated frequency of the ith allele/haplotype (Nei, 1987). We used the derived population parameter θH as another expression of the intrapopulation diversity (Watterson, 1975). The population mutation parameter, θH (Watterson estimator) is equal to θH=[h/1−h]=2Nμ and depends on N being the effective number of chromosomes and μ the mutation rate. The θH=[h/1−h]=2Nμ for the sex chromosomes is equal to θH(Y)=2NYμY and θH(X)=2NXμX. According to the different number of chromosomes in each sex, NY=Nmales and NX=2Nfemales+Nmales; the ratio θH(Y)/θH(X) in each population depends on the sex-specific effective population sizes and on the Y/X mutation rate ratio.
Estimates of interpopulation diversity
We used ARLEQUIN version 2.0 software (Schneider, 2000) to perform analyses of molecular variance (AMOVA). AMOVA produces estimates of variance components reflecting the correlation of allelic/haplotypic diversity at different levels of hierarchical subdivision. This included (i) FCT: cluster of subpopulations relative to total population, (ii) FSC: subpopulation relative to cluster of subpopulations and (iii) FST: subpopulation relative to total population. Significance levels of the variance components and the corresponding F-statistics were obtained by comparison of the actual values with the distribution of 10 000 values obtained by randomization.
Estimates of sex-specific processes
The global differentiation coefficients FST(X) and FST(Y) were computed and compared to estimate the relative impact of sex-specific processes (migration rate and effective population size) that shape diversity patterns for the two sexes.
Assuming an island model with neutrality, the theoretical models predict for a given genetic system that FST=1/(1+2ν), where ν=Nm (Wright, 1951), with N the effective number of chromosomes and m the rate of migration. For the X- and Y-chromosomes, we have, respectively, FST(X)=1/(1+2νX) and FST(Y)=1/(1+2νY), with N(Y) the effective number of Y chromosomes, N(X) the effective number of X-chromosomes, m(X) the rate of X-chromosome migration and m(Y) the rate of Y-chromosome migration.
Then, from these differentiation coefficients FST(X) and FST(Y), we can calculate the νX/νY ratio as follows:

Using equation (1), in two specific cases, we can evaluate to what extent the migration rate and the effective population size shape the genetic differentiation across all populations.
Hypothesis 1 (H1):
-
Male and female effective population sizes are equal (Nfemale=Nmale=N), then N(X)=3N(Y); hence, νX/νY depends only on different migration rates of males and females. As
 , the ratio of male to female migration rate is given by equation (2):
, the ratio of male to female migration rate is given by equation (2): 
 , the ratio of male to female migration rate is given by equation (2):
, the ratio of male to female migration rate is given by equation (2): 
Hypothesis 2 (H2):
-
Male and female migration rates are equal (mfemale=mmale=m); hence, m(X)=m(Y)=m, in which case the ratio νX/νY depends only on different male and female effective population sizes. According to Hartl and Clark (1989), NX=9(NfemaleNmale)/2(Nmale+Nfemale), so the male to female effective population size ratio is given by equation (3):


Testing correlation between X- and Y-derived distance matrices
We computed FST genetic distance matrices between populations for the X- and Y-linked markers and compared them using the Mantel test (Mantel, 1967). The significance of the correlation between the two matrices was determined by random permutations (n=1000) and measured by the Z-statistic (Manly, 1997). The computation was performed using the Microsoft Excel macro Mantel.xla v. 1.2 (compiled by Dr RA Briers – r.briers@shef.ac.uk).
Results
X- and Y-linked gene diversity: estimates of θH(Y)/θH(X) ratio
The mean values of DYS413a,b and DXS8175 haplotype/gene diversity for each group of populations are reported in Table 1. We calculated an estimate of the mean and the variance of the θH(Y)/θH(X) ratio for each group of populations considered. Averaging, the θH(Y)/θH(X)= 1.7951.495 and this ratio varies drastically between and within each geographic area considered (Table 1). Extreme values are reached in Asia (3.6622.005) and in West Africa (0.5110.291).
Distribution of the genetic variability within and between populations: analysis of molecular variance and genetic distances
Y-linked genetic variation
The overall FST value for all 33 populations is 0.194 (P<0.001), indicating that a large proportion of the overall Y-chromosomal variation (80.6%) results from intrapopulation differences (compared to the X data here, Table 2). Overall FST values calculated for all populations of Africa, Europe and Asia are, respectively, 0.240, 0.068 and 0.032, reflecting a larger genetic differentiation of African populations than in other groups. The North vs South Sahara populations show a high degree of differentiation (FCT=0.184, P<0.01) and these two groups seem heterogeneous (FSC=0.172, P<0.001). A very similar pattern is observed when eastern and western African populations are grouped (FCT=0.160, P<0.04; FSC= 0.147, P<0.001). In West Africa, the FST value is 0.174 (P<0.001), the lowest value found in Africa and lower than the average Y-linked differentiation between all populations (0.194). This reflects a lower genetic differentiation between these populations in this geographic area. In Europe, no genetic differentiation appears by grouping Northern and Southern populations (FCT=−0.013, NS).
X-linked genetic variation
The overall FST value calculated for all populations is 0.041 (P<0.001), indicating that most of X-chromosomal variation results from intrapopulation differences (96%, Table 2). This tendency remains unchanged when we consider populations from Africa (FST=0.034, P<0.001), Europe (FST=0.002, NS) or Asia (FST=−0.014, NS). North vs South Sahara populations show the highest FST value in Africa (FST=0.04, P<0.001), suggesting a high degree of genetic differentiation between these two groups (FCT=0.03, P<0.01). This kind of genetic structuring cannot clearly see between western and eastern groups in Africa (FCT=−0.006, NS). The West African populations show a low level of genetic differentiation (FST=0.009, P<0.01). In Europe, no genetic differentiation is observed between groups from the North and South (FCT=0.004, NS).
Testing correlation between X- and Y-derived distance matrices
We compared pairwise FST distance matrices for the X- and Y-linked markers by the Mantel test. We find a lack of association between these matrices (r=−0.023; z=0.065), which confirms the discordant pattern of genetic variation between X and Y-linked homologous markers in the AMOVA analyses.
Estimates of sex-specific parameters inducing differential genetic differentiation of populations
Considering a neutral model in which FST=1/(1+2ν), with 2ν=2Nm and applying this model to X- and Y-chromosomes, we used equations (1), (2) and (3) to evaluate both the sex-specific effective population size and migration rate ratios. Global genetic differentiation values of FST(X)=0.03 and FST(Y)=0.19 have been input in equation (1) and yielded a ratio νY/νX=7.58.
Under H1 (Nfemale=Nmale=N), this yields m(X)/m(Y)=2.53 and mfemale/mmale=3.29. Under this hypothesis, the migration rate should be three-fold higher for females than for males to explain the global diversity pattern.
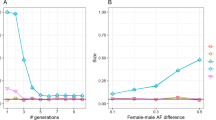
Under H2 (mfemale=mmale=m), thus N(X)/N(Y)=7.58 and Nfemales/Nmales=10.71. Under this hypothesis, the effective population size of females should be 10-fold higher than that of males to explain the results. To provide a wider picture of the variation of these sex-specific processes among populations, we show the wide range of scenarios compatible with our X- and Y-genetic distances in Figure 3.

(a) The curve represents all potential scenarios compatible with an average genetic differentiation between all populations corresponding to FST(X)=0.03 and FST(Y)=0.19. The scenario corresponding to each geographic area has been deduced from the gene diversity values. (b) Details of the general curve with most of the scenarios involving sex-specific processes explaining X- and Y-linked genetic differentiation between populations are seen.
Inference of sex-specific processes using intrapopulation information
In order to reconcile the X- and Y-linked data under the neutral island model considered above, the migration rate of females must be three times higher than that of males (given equal effective population sizes), or the effective population size of females must be 10 times higher than that of males (given equal migration rates).
According to Slatkin (1985), in a structured system with symmetric migration, the migration has an important impact only on the interpopulation differentiation, but a much smaller impact on the intrapopulation diversity. Assuming symmetric migration, we consider the theoretical case in which the 33 populations studied should be at equilibrium, to explain the variation of θH(Y)/θH(X) observed among populations. We know that θH(Y)/θH(X) depends on the effective population size (N) and the mutation rate (μ). If we assume that μ(Y)/μ(X)=6.6 (see above), we can explain the different θH(Y)/θH(X) observed between populations by the Y/X effective population sizes as follows: taking the mean value of θH(Y)/θH(X) in all populations to be 1.79 (see Table 1) and μ(Y)/μ(X)=6.6, 6.6NY/NX=1.79. We found that NX=3.68NY, corresponding to an Nfemale/Nmale=1.38. Assuming that this value is close to 1 (equal male and female effective population sizes), we can deduce from NX=3.68NY and from equation (2), the corresponding male to female migration rate ratio: (mX/mY)=7.58/3.68=2.06 and  .
.
Then, for similar male and female effective population sizes, a μ(Y)/μ(X)=6.6 and θH(Y)/θH(X)=1.79, the expected migration rate of females must be 2.59 times higher than that of males to reconcile differences in X- and Y-linked genetic variation. We positioned on Figure 3 all the values of θH(Y)/θH(X) calculated for the eight groups of populations considered. This shows that a wide range of migration and demographic scenarios is compatible with our data and can be invoked to explain the drastic variation of θH(Y)/θH(X) between the different geographical areas (from Mfemale/Mmale=1.43 and Nfemale/Nmale=3.77 in South Africa to Mfemale/Mmale=5.89 and Nfemale/Nmale=0.49 in Asia). The outlying value for the western African populations suggests an opposite scenario, involving a higher migration rate or effective population size for males compared to females.
Discussion
Few studies have described the genetic variability of XY-homologous markers in human populations (Scozzari et al, 1997; Karafet et al, 1998; Kotliarova et al, 1999; Carvalho-Silva and Pena, 2000) compared to those studying mitochondrial and Y-chromosomal markers (eg Seielstad et al, 1998; Jorde et al, 2000). The usefulness of XY-homologous markers studied so far has been compromised by the technical problem that the X- and Y-linked copies cannot be individually amplified. Coamplification limits the use of classic estimators for inferring the features of intra- and interpopulation differentiation (eg Karafet et al, 1998). In this paper, we used the DXS8175 and DYS413a,b homologous microsatellites, which can be specifically amplified from the X- and Y-chromosomes.
Higher Y- than X-linked genetic differentiation: possible and testable explanations
Our findings, based on a data set comprising 33 populations from Europe, Asia and Africa, reveal a clear discordant pattern of genetic variation between X- and Y-homologous markers for all geographical areas considered, with between-population differentiation always greater for the Y- than for the X-chromosome. Two hypotheses could explain this result: sex specific processes or differential selection. We consider these in turn.
Sex- and population-specific processes?
Sex-specific processes could involve a lower migration rate of males compared with females, due to patrilocality, and/or a lower effective population size for males compared with females due to demographic processes.
In order to test the impact of such processes, we compared X- and Y-linked F-statistics calculated including all populations. However, a given F-statistics value is compatible with many different scenarios. We next analyzed the variability within each population for X- and Y-linked markers using Watterson's estimator θ. By comparing FST(X) and FST(Y) values (assuming a simple 1/1+4Nm), we showed that if all the populations considered in the present study were at equilibrium and exchanged migrants symmetrically, the X/Y genetic differentiation could be explained by sex-specific migration processes (mfemale/mmale=3.29), by sex-specific demographic processes (Nfemales/Nmales=10.71) or by a combination of both.
It seems unrealistic to draw conclusions using a simple and unique scenario involving the different sex-specific processes. In general, our data showed that a lower migration rate of males compared with females (due to the tendency for a female to move into her husband's natal household) and a lower effective population size for males compared with females (such as the prevalence of polygyny over polyandry, or cultural practices) could be two important factors explaining genetic structuring in human populations (eg Salem et al, 1996; Bamshad et al, 1998; Sibert et al, 2002). This general tendency may be reversed in some populations: as we showed in West Africa, the results suggest an opposite scenario (including populations originating from the Bantu expansions). A similar result has been reported by Hammer et al (2001) and Wood et al (2005) providing the evidence that the different approaches converge.
The large fluctuation of the Watterson's estimator ratio θH(Y)/θH(X) between populations in the same region suggests that these sex-specific features may be specific to each population and that they may vary whatever the geographical scale considered. Variations due to local cultural practices (including language affinities between populations) are consistent with these findings (Oota et al, 2001; Hamilton et al, 2005; Wood et al, 2005).
The ratio of the Y/X substitution rates used in our calculations has a major influence on our estimates of sex-specific parameters. This value, 6.6, is higher than previously reported values, which vary from 2.2 to 2.8 in higher primates (Li et al, 2002). The two Y-linked sequences studied here lie within each arm of a ‘palindrome’ – large highly similar repeat units arranged ‘tail-to-tail’ (Figure 2); gene conversion processes are known to be highly active between such sequences on the Y-chromosome (Rozen et al, 2003), and could act (as they do in the direct repeats flanking the AZFa region; Bosch et al, 2004) to enhance interspecific sequence divergence.
Sex-specific selection?
In assessing the potential impact of differential selection on genetic markers, we need to consider both direct effects on the sequences under consideration, and indirect effects due to linkage.
We have assumed a minimal impact of differential selection acting directly on the XY-homologous markers studied here, because of the similarity in genomic context (Balaresque et al, 2003), and despite the 50 million years separating each from a common ancestor (Lahn and Page, 1999). Although there is no reason to expect selective pressures to act directly on microsatellites in noncoding regions, the low sequence divergence of flanking sequences among primate species suggests that selection may be playing a role (Balaresque et al, 2003). The VCX and VCY genes are in close proximity to the microsatellites, but given that they appear to be under similar selective pressures (Lahn and Page, 1999), this should result in equivalent selective bias at both the X and Y loci.
Indirect selection on the microsatellites is affected by the recombination behavior of the sex chromosomes. As the Y-chromosome is largely nonrecombining, it represents only a single realization of the evolutionary process, and selection acting anywhere along its length could affect the diversity of DYS413a,b. However, this problem is unavoidable in any study, whether it compares the Y with the X, or with mtDNA. Selection could also be affecting DXS8175 through linkage to a locus under selection on the X-chromosome; however, because of recombination in females, the X-chromosome represents not one but many genealogies, with different selective histories.
The ideal marker for examining sex-specific phenomena (uniparentally inherited through males and females, but having similar molecular characteristics) does not exist. However, a combination of Y-chromosomal and mitochondrial haplotyping with the analysis of XY-homologous loci may prove to be powerful tool. A large potential resource of XY-homologous markers exists and could be readily accessed by simple in silico searches. Analysis of additional markers mapping to different regions of the sex chromosomes would obviate the effects of selection on individual X-linked loci, and would also allow the use of single-copy Y-specific loci, without the complicating factor of gene conversion processes.
References
Balaresque P, Toupance B, Heyer E, Crouau-Roy B (2003). Evolutionary dynamics of duplicated microsatellites shared by sex chromosomes. J Mol Evol 57: 1–10.
Balaresque P, Toupance B, Quintana-Murci L, Crouau-Roy B, Heyer E (2004). Sex-specific selection on the human X chromosome? Genet Res Camb 83: 1–8.
Bamshad MJ, Watkins WS, Dixon ME, Jorde LB, Rao BB, Naidu JM et al (1998). Female gene flow causes genetic stratification of the Hindu cast system. Nature 395: 651–652.
Bosch E, Hurles ME, Navarro A, Jobling MA (2004). Dynamics of a human interparalog gene conversion hotspot. Genome Res 14: 835–844.
Carvalho-Silva DR, Pena SD (2000). Molecular characterization and population study of an X chromosome homolog of the Y-linked microsatellite DYS391. Gene 247: 233–240.
Chen FC, Li WH (2001). Genomic divergences between humans and others hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68: 444–456.
Cummins J (2001). Mitochondrial DNA and the Y chromosome: parallels and paradoxes. Reprod Fertil Dev 13: 533–542.
Hamilton G, Stoneking M, Excoffier L. (2005). Molecular analysis reveals tighter social regulation of immigration in patrilocal populations than in matrilocal populations. Proc Natl Acad Sci USA 102: 7476–7480.
Hammer MF, Karafet TM, Redd AJ, Jarjanazi H, Santachiara-Benerecetti S, Soodyall H et al (2001). Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 18: 1189–1203.
Hartl DL, Clark AG (1989). Principles of Population Genetics, 2nd edn. Sinauer Associates Inc: Sunderland, MA.
Jorde LB, Watkins WS, Bamshad MJ, Dixon ME, Ricker CE, Seielstad MT et al (2000). The distribution of human genetic diversity: a comparison of mitochondrial, autosomal, and Y-chromosome data. Am J Hum Genet 66: 979–988.
Karafet T, de Knijff P, Wood E, Ragland J, Clark A, Hammer MF (1998). Different patterns of variation at the X- and Y-chromosome-linked microsatellite loci DXYS156X and DXYS156Y in human populations. Hum Biol 70: 979–992.
Kayser M, Brauer S, Weiss G, Schiefenhovel W, Underhill P, Shen P et al (2003). Reduced Y-chromosome, but not mitochondrial DNA, diversity in human populations from West New Guinea. Am J Hum Genet 72: 281–302.
Kotliarova SE, Toda T, Takenaka O, Matsushita I, Hida A, Shinka T et al (1999). Novel (CA)n marker DXYS241 on the nonrecombinant part of the human Y chromosome. Hum Biol 71: 261–275.
Lahn BT, Page DC (1999). Four evolutionary strata on the human X chromosome. Science 286: 964–967.
Li WH, Yi S, Makova K (2002). Male-driven evolution. Curr Opin Genet Dev 12: 650–656.
Malaspina P, Ciminelli BM, Viggiano L, Jodice C, Cruciani F, Santolamazza P et al (1997). Characterization of a small family (CAIII) of microsatellite-containing sequences with X–Y homology. J Mol Evol 44: 652–659.
Manly BFJ (1997). Randomization, Bootstrap and Monte Carlo Methods in Biology. Chapman & Hall: New York.
Mantel N (1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
Mathias N, Bayes M, Tyler-Smith C (1994). Highly informative compound haplotypes for the human Y chromosome. Hum Mol Genet 3: 115–123.
Nei M (1987). Molecular Evolutionary Genetics. Columbia University Press: New York.
Oota H, Settheetham-Ishida W, Tiwawech D, Ishida T, Stoneking M (2001). Human mtDNA and Y-chromosome variation is correlated with mutrilocal versus patrilocal residence. Nat Genet 29: 20–21.
Perez-Lezaun A, Calafell F, Comas D, Mateu E, Bosch E, Martinez-Arias R et al (1999). Sex-specific migration patterns in Central Asian populations, revealed by analysis of Y-chromosome short tandem repeats and mtDNA. Am J Hum Genet 65: 208–219.
Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D. et al (2005). The DNA sequence of the human X chromosome. Nature 434: 325–337.
Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterson RH et al (2003). Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423: 873–875.
Salem AH, Badr FM, Gaballah MF, Paabo S (1996). The genetics of traditional living: Y-chromosomal and mitochondrial lineages in the Sinai Peninsula. Am J Hum Genet 59: 741–743.
Schneider S (2000). Arlequin: A Software for Population Genetics Data Analysis. Genetics and Biometry Laboratory, University of Geneva: Switzerland.
Scozzari R, Cruciani F, Malaspina P, Santolamazza P, Ciminelli BM, Torroni A et al (1997). Differential structuring of human populations for homologous X and Y microsatellite loci. Am J Hum Genet 61: 719–733.
Seielstad MT, Minch E, Cavalli-Sforza LL (1998). Genetic evidence for a higher female migration rate in humans. Nat Genet 20: 278–280.
Sibert A, Austerlitz F, Heyer E (2002). Wright–Fisher revisited: the case of fertility correlation. Theor Popul Biol 62: 181–197.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG et al (2003). The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837.
Slatkin M (1985). Gene flow in natural population. Annu Rev Ecol Syst 16: 393–430.
Wilder JA, Kingan SB, Mobasher Z, Pilkington MM, Hammer MF (2004). Global patterns of human mitochondrial DNA and Y-chromosome structure are not influenced by higher migration rates of females versus males. Nat Gent 36: 1122–1125.
Watterson GA (1975). On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7: 256–276.
Wood ET, Stover DA, Ehret C, Destro-Bisol GD, Spedini G, McLeod H et al (2005). Contrasting patterns of Y chromosome and mtDNA variation in Africa: evidence for sex-biased demographic processes. Eur J Hum Genet 13: 867–876.
Wright S (1951). The genetical structure of populations. Ann Eugenic 15: 323–354.
Acknowledgements
We thank Mark Jobling for his advices and assistance with the manuscript; F Austerlitz for his help in data analysis; A Chaventre, GF De Stefano, A Baali, M Cherkaoui, G Bellis, P Kouam, A Pitte, MS Isaad, G Larrouy and A Sevin for providing DNA samples; R Jambou-Clerc for his technical assistance and J Sloggett, N Poulet, B Massonnet and M Lathuillire for fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Balaresque, P., Manni, F., Dugoujon, J. et al. Estimating sex-specific processes in human populations: Are XY-homologous markers an effective tool?. Heredity 96, 214–221 (2006). https://doi.org/10.1038/sj.hdy.6800779
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800779
Keywords
This article is cited by
-
The Cocksure Conundrum: How Evolution Created a Gendered Currency of Corporate Overconfidence
Adaptive Human Behavior and Physiology (2022)
-
Differential pattern of genetic variability at the DXYS156 locus on homologous regions of X and Y chromosomes in Indian population and its forensic implications
International Journal of Legal Medicine (2013)
-
Molecular sexing and population genetic inference using a sex-linked microsatellite marker in the nine-spined stickleback (Pungitius pungitius)
BMC Research Notes (2011)
