
Abstract
Bedouin are traditionally nomadic inhabitants of the Persian Gulf who claim descent from two male lineages: Adnani and Qahtani. We have investigated whether or not this tradition is reflected in the current genetic structure of a sample of 153 Bedouin males from six Kuwaiti tribes, including three tribes from each traditional lineage. Volunteers were genotyped using a panel of autosomal and Y-STRs, and Y-SNPs. The samples clustered with their geographical neighbours in both the autosomal and Y-chromosomal analyses, and showed strong evidence of genetic isolation and drift. Although there was no evidence of segregation into the two male lineages, other aspects of genetic structure were in accord with tradition.
Similar content being viewed by others

Introduction
Kuwait lies in the Arabian Peninsula at the head of the Persian Gulf. Although it is located near a possible migration route out of Africa that may have been used over 50 000 years ago (Jobling et al., 2004), such early events seem to have had little impact on the current populations of Kuwait and the first known settlement dates only to the third century BCE when the Ancient Greeks colonized the island of Failaka under Alexander the Great. Historians believe that the current settlement was established between the late 16th and 18th centuries CE (Dickson, 1956). The population of Kuwait is divided into two main groups: townspeople and Bedouin tribal groups ( Qaba'il) with affiliations to their ‘cousins’ (
Qaba'il) with affiliations to their ‘cousins’ ( wild ‘am) who inhabit other parts of the Arabian Peninsula. The Bedouin are not geographically isolated from each other and are traditionally nomadic, moving from one region to another seeking pasture for their herds to graze and water for survival (Dickson, 1967). The Bedouin conventionally claim descent from two main lineages: from Adnan son of Ishmael—the Adnani lineage—or from Qahtan (or Joktan)—the Qahtani (or Joktani) lineage.
wild ‘am) who inhabit other parts of the Arabian Peninsula. The Bedouin are not geographically isolated from each other and are traditionally nomadic, moving from one region to another seeking pasture for their herds to graze and water for survival (Dickson, 1967). The Bedouin conventionally claim descent from two main lineages: from Adnan son of Ishmael—the Adnani lineage—or from Qahtan (or Joktan)—the Qahtani (or Joktani) lineage.
We wished to investigate the genetic structure of the Kuwaiti Bedouin and consider how they compare with neighbouring populations, what the genetic consequences of their traditional lifestyle might be, and whether the social lineages that identify their descent groups are also reflected in their genetic lineages.
The non-recombining part of the Y chromosome was chosen for analysis. Its high levels of population differentiation—a consequence of its low effective population size and, sometimes, of patrilocality—make it generally highly informative regarding genetic structure and differentiation (Jobling and Tyler-Smith, 2003). In addition, its strict paternal inheritance allows comparisons to be made between traditional male-line descent groups and observed male-line genetic structures. It is a single locus, however, and conclusions based on it may not be representative of the rest of the genome. We, therefore, complemented the Y-chromosome analysis with that of a widely used panel of autosomal STRs (Collins et al., 2004).
Samples for DNA analysis were collected from volunteers from the Adnani tribes of Al-Aniza, Mutran and Awazim (a Suluba tribe), and the Qahtani tribes of Ajman, Shimar and Murrah. This selection is representative of the most prominent tribes of Adnani and Qahtani lineages of Arabia. The results from the sample were compared with data from other populations derived from the literature.
Materials and methods
Samples
One hundred and fifty-three distantly related male subjects—known not to share a common ancestor within the last three generations—were collected from six Bedouin tribes. Ethical review was completed by a research ethics committee of The University of Sheffield (UK) and The Kuwait University ethical review board (Kuwait). Informed consent was obtained from all participants. Specimens were collected as buccal swabs. The Bedouin were from the two traditional lineages—Adnani: Mutran (n=30), Aniza (n=21) and Awazim (n=37), and Qahtani: Ajman (n=39), Shimar (n=21) and Murrah (n=5; because of the small sample size, Murrah specimens were used only in the autosomal analysis). Samples were collected from four main Kuwaiti Governorates: Awazim and Aniza tribes from the Al-Assima (capital) Governorate (29.4 Lat, 42.2 Long.), Ajman and Murrah from the Al-Ahmadi Governorate (28.7 Lat, 48.3 Long.), the Mutran sample from the Al-Farwaniyah Governorate (29.2 Lat, 47.9 Long.) and the Shimar sample from both the Al-Jahrah (29.3 Lat, 47.6 Long.) and Al-Ahmadi Governorates. Locations are shown in Supplementary Figure 1. Kuwaiti Bedouin do not marry outside their own tribe and samples collected in the capital are not admixed. DNA was extracted from buccal cells using the BuccalAmp DNA Extraction Kits and protocol (Epicentre, Madison, WI, USA).
Autosomal STR typing
Amplification with the identifiler multiplex was performed directly on 5 μl (0.1 ng μl–1) of the DNA extract using a 12.5-μl reaction volume following the recommendations of the manufacturer (Collins et al., 2004) on a GeneAmp PCR System 2400 (Applied Biosystems, Foster City, CA, USA). PCR cycle conditions were set at 95 °C for 11 min; 28 cycles: 94 °C for 1 min, 59 °C for 1 min, 72 °C for 1 min; then 60 °C for 60 min. The amplified products were electrophoretically separated on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems). After separation, the fluorescent fragments were analysed using GeneMapper-ID version 3.1 (Applied Biosystems).
Y-STR typing
Amplification with the Y-filer multiplex was performed directly on 5 μl (0.1 ng μl–1) of the DNA extract using a 12.5-μl reaction volume following the recommendations of the manufacturer (Mulero et al., 2006) on a GeneAmp PCR System 2400 (Applied Biosystems). PCR conditions were 95 °C for 11 min; 30 cycles: 94 °C for 1 min, 61 °C for 1 min, 72 °C for 1 min; then 60 °C for 80 min. The amplified products were electrophoretically separated on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems). After separation, the fluorescent fragments were analysed using GeneMapper-ID version 3.1 (Applied Biosystems). Y-STR typing was carried out according to the manufacturer's protocol.
Binary polymorphism typing
Y-SNP typing was carried out with a 96 Y-SNP kit (version 2.0) manufactured by Marligen Biosciences (Ijamsville, MD, USA). The kit comprised a core detection kit (Haplogroups A-R), a broad screening kit and 11 individual genotyping kits (Supplementary Table 1). Y-SNP fragments were amplified by multiplex PCR, in which 1–10 μl (1 ng μl–1) of each DNA sample was used to amplify the Y-chromosomal DNA. Amplified DNA was labelled with a fluorescent tag. The labelled products were hybridized to Y-SNP-specific oligonucleotides immobilized onto individually colour-coded beads. The Luminex 100 System (Luminex Corporation, Austin, TX, USA) was used to read the fluorescence of the products. Binary polymorphism typing was carried out according to the manufacturer's protocol at Marligen Biosciences (Ijamsville).
Data analysis
Analysis of molecular variance (AMOVA) was carried out with Arlequin version 2.0 (Schneider et al., 2000). AMOVA measures the proportions of variance found within and between populations or groups of populations. Y-STR variation was used to calculate population pairwise RST values. Similarly, SNP variation was used to calculate pairwise φST values. In addition, autosomal allele frequencies were used to calculate FST pairwise-genetic distances. The distance matrix used consisted of the number of steps by which each pair of STR haplotypes or SNP haplogroups differed. Multidimensional scaling (MDS) plots were constructed using SPSS version 12.0.
STRUCTURE 2.1 (Pritchard et al., 2000) was used to infer clusters of individuals on the basis of the genetic data from unlinked loci alone. The model assumes the presence of K populations (where K is unknown and is specified in advance for each run of the program). Allele frequencies at each locus characterize each assumed K. For all simulations and calculations, a no-admixture model was assumed, with allele frequencies correlated among groups. In the no-admixture model, each individual comes from one of the K populations—that is populations do not have mixed ancestry; anticipating that the volunteers are either of a Qahtani or Adnani ancestry. The program was run twice, once with K=1–6 as the predetermined number of Bedouin populations, and once comparing the Bedouin sample to worldwide populations also predetermined at K=1–6. Both runs used 50 000 iterations for the burn-in period and 100 000 additional iterations to obtain parameter information. Each run was carried out five times and posterior probabilities for each K were computed for each set of runs.
Median-joining networks were constructed using Network version 4.1.1.2 or 4.5.1.0 (Bandelt et al., 1999). A weighting method with a four-fold range was used in the construction of the networks, with specific weights assigned for each STR locus (Qamar et al., 2002) taking into account the Y-STR variation across the Bedouin samples. The network for haplogroup J contained many high-dimensional reticulations. This was resolved by applying the reduced median followed by median-joining network methods.
Bayesian Analysis of Trees With Internal Node Generation (BATWING) (Wilson et al., 2003) was used to estimate population-genetic parameters and the time to most recent common ancestor (TMRCA) of a set of chromosomes. The model assumes the single-step mutations of STRs and the demographic model chosen was exponential growth from an initially constant-sized population, with or without subdivision in different runs of the program. All 15 unique Y-STR loci and all variable Y-SNP markers were used. A generation time of 25 years was used as appropriate for the mutation rate estimated by Zhivotovsky et al. (2004) and additional parameters were as described earlier (Xue et al., 2006). The 95% confidence intervals of posterior values for the TMRCA, effective population size (Nposterior) and expansion time were determined from the program's output.
Results
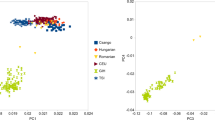
We first investigated the genetic relationships revealed by genomewide autosomal markers among the Bedouin and 14 other populations taken from the literature (Supplementary Table 2) by computing the pairwise FST genetic distances (Supplementary Table 3) from the allele frequencies of 13 STR CODIS loci (Figure 1a). Overall, with the exception of the Aniza tribe, the Bedouin clustered with the Arabic-speaking populations of the Middle East, and were distinct from Far Eastern and European populations. However, although the populations of the Persian Gulf clustered closely together, the Bedouin populations formed a much looser part of the cluster, most marked for the Aniza.

Autosomal STR analyses of Kuwaiti Bedouin and additional populations. (a) MDS plot of population-pairwise genetic distances (FST values). (b) Estimated population structure based on the highest probability STRUCTURE run at K=3. Each individual is represented by a thin vertical line, which is partitioned into K coloured segments that represent the individual's estimated membership fractions in each of the K clusters. Populations are shown below the figure.
To complement this analysis, we explored the genetic clustering identified by the program STRUCTURE. There was no evidence of the presence of significant genetic structure within the Bedouin corresponding to Adnani and Qahtani clusters. The model with the highest posterior probability value was K=1 (ln P(D)=−7953 for K=1 compared with −8037 for K=2; Table 1), implying that the genetic data favoured a single cluster containing all the Bedouin populations examined.
The same program was used to test for the presence of broader genetic structure when Bedouin populations were compared with a worldwide population data set (Supplementary Table 2). In the worldwide comparison, a model containing three clusters was favoured (at K=3 ln P(D)=−42339; Table 1). In this analysis, almost all individuals showed membership of multiple clusters, but some populations displayed membership of predominant clusters that corresponded to geographical affiliation. For example, Adima Muslim Egyptians (63%) and Coptic Egyptians (65%) showed a distinct ‘North African’ cluster (Figure 1b, red colour). Another ‘European’ cluster was observed among a high proportion of the South Belgians (56%, blue colour). Correspondences between clusters and populations were less clear for the Iraqi and Qatari samples; however, individuals displayed partial membership of multiple clusters with some populations showing identities that did not correspond to geographic, linguistic or ethnic affiliation. It was noticeable that all of the Bedouin tribes with the exception of Aniza belonged predominantly to a single cluster (cluster 2, green colour). The Aniza, similar to the samples drawn from Qatar and Iraq, were distributed among three clusters (Supplementary Table 4).
The autosomal analyses thus indicated that the Bedouin populations resembled their geographical neighbours, but showed larger inter-population-genetic distances, perhaps reflecting high levels of genetic drift, and that these tendencies were particularly marked in the Aniza.
We next analysed the Y-chromosomal lineages. Fifteen-locus Y-STR haplotypes were obtained for all individuals (n=148, the Murrah were excluded; Supplementary Table 5). Table 2 shows the Y-haplotype diversities observed in the five Bedouin samples studied and compared with published data from other populations. Few studies have reported 15-locus haplotypes, so it was necessary to base broader comparisons on nine loci (Supplementary Table 2). The Bedouin populations stand out even in this simple analysis because of their strikingly low diversity. With 15 loci, the Bedouin show the lowest three diversity values. With nine loci, the Bedouin population diversities were again unusually low, similar to some other Persian Gulf samples and with lower values seen only in Zriba, Berber and Somali populations.
Y-SNP analysis classified the Bedouin samples into nine haplogroups (Table 3, Supplementary Figure 2). Haplogroup diversities were also low, ranging from zero for the Ajman—all members belonged to haplogroup J1—to 0.6 for the Aniza (Table 3). Haplogroup J1 was seen at very high frequency in the combined sample: ∼84%, or 124 individuals, and was present at >50% in all individual populations. The other haplogroups identified were R1a1 (at 6.75%), E3b3 (6.00%), G2 (3.40%), R1b3 (1.35%) and K2, E3b1, Q* and R2 (each 0.67%).
The patrilineal genetic relationships between the populations were examined using Y-STR or Y-haplogroup frequency data to calculate pairwise-genetic distances (φST) between populations, and displayed in two dimensions using MDS plots (Figure 2). The MDS plots showed the same general features as the autosomal plot (Figure 1a): clustering with their geographical neighbours, but with larger distances between the Bedouin samples. The Aniza, however, did not stand out in the Y-chromosomal analyses. We investigated the Y-chromosomal-genetic structure further by carrying out an AMOVA analysis with either Y-STRs or Y-SNPs. The results were similar with the two sets of markers (Table 4), so are discussed together. When populations were grouped according to continent, a large fraction of the variation lay between groups (23% with STRs, 15% with SNPs; P<0.0001) reflecting the high degree of geographical structure also found in many other studies (Jobling et al., 2004). When the Bedouin populations were analysed alone, substantial variation was found between populations. At 18% for Y-SNPs, this exceeded the variation between continental groups—again illustrating the large genetic distances between these populations. When the Bedouin samples were grouped into Adnani and Qahtani lineages, however, no variation was found between the groups (Table 4).

Y-chromosomal analyses of Kuwaiti Bedouin and additional populations. (a) MDS plot based on population-pairwise genetic distances from Y-STRs (RST values). (b) MDS plot based on population pairwise-genetic distances from Y-SNPs. Population names are shown in each section.
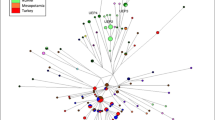
To investigate the phylogenetic relationships between the Y-STR haplotypes in the Bedouin and other samples, median-joining networks were constructed. The haplogroup J1 network, containing 84% of the chromosomes, is illustrated in Figure 3. It shows a high-frequency central haplotype present in most of the populations examined, both Bedouin and non-Bedouin, and a starlike structure with many radiating branches. Although individual haplotypes in the branches are often shared between populations, some clustering is evident, such as the Ajman (yellow) haplotypes in the upper right part of the network. The TMRCA—estimated using the assumption that the central haplotype is ancestral—was 3700 years.

Median-joining network of haplogroup J1 Y-STR haplotypes. Circles represent haplotypes with an area proportional to frequency and are coloured to indicate the population of origin. Lines represent mutational differences between the haplotypes.
Modelling of the demography of the individual populations using BATWING revealed median Nposterior estimates (see Table 5) ranging from 560 (Ajman) to 2300 years (Shimar) and median expansion times from 2600 (Awazim) to 8500 years (Shimar). These expansion times are much more recent than those found using the same approach in other parts of the world (Xue et al., 2006) and point to an unusual demography for the Bedouin. The population TMRCA estimate for the Ajman sample is very recent (median 4300 years) because of the presence of a single haplogroup, and comparable to the J1 network TMRCA. TMRCAs for the other populations reflect the coalescence of the Y lineages from different haplogroups before the formation of the Bedouin populations.
Discussion
Although earlier studies have investigated classical genetic (Singh Sawhney et al., 1984; Al-Hilli, 1985; Al-Bustan et al., 2002) and molecular (Al-Nassar et al., 1981; al-Nassar et al., 1995; al-Nassar et al., 1996; Sebetan and Hajar, 1998) markers, including Y-chromosomal markers (Nebel et al., 2001) in Bedouin populations, and one Bedouin sample from Israel is present in the HGDP panel (Cann et al., 2002), the Kuwaiti Bedouin have thus far been little studied. They share some Y-chromosomal features with the Negev Bedouins (Nebel et al., 2001): a high frequency of haplogroup J1 and low STR diversity within it, but none of the Kuwaiti individuals sampled carries the Bedouin Modal Hapolotype and thus their recent genetic history is distinct. Four main conclusions now emerge from our work:
First, the Kuwaiti Bedouin populations resemble their geographical neighbours. The autosomal markers used in this study were chosen for their ability to discriminate between individuals within any population, and this property, together with their small number, leads to high levels of noise when they are used to compare populations. Nevertheless, they reveal the genetic relationships between the Bedouin and other populations from the Arabian Peninsula (Figure 1), as expected from our understanding of the history of the region. The same conclusion is obtained with Y-chromosomal markers (Figure 2).
Second, the Bedouin exhibit evidence of small effective population sizes and substantial genetic drift. This is reflected in the low within-population diversity values for Y-chromosomal loci, and the low estimates of effective population size with BATWING; note that the latter values are estimates of the size when growth began—that is ∼3–9000 years ago—and so are exceptionally low. Genetic drift leads to large distances between populations and these are seen both in the autosomal analyses—most marked for the Aniza (Figure 1a)—and the Y-chromosomal analyses (Figure 2). Again, these are exactly the genetic consequences expected from their lifestyle as small groups of desert dwellers.
Third, no subdivision was seen according to the traditional Adnani and Qahtani lineages. This is illustrated by the lack of STRUCTURE grouping into more than one cluster (Table 1), interspersion of populations belonging to the traditional lineages in the autosomal and Y-chromosomal MDS plots (Figures 1 and 2) and lack of distinction between the lineages in the AMOVA analyses (Table 4). This finding is contrary to historical and socio-cultural expectations and could be explained in a number of ways. For example, a common genetic pattern could have been established in an ancestral population and retained by the two groups; alternatively, there could have been sufficient gene flow between the tribes to prevent genetic differences building up; or each individual population could have diverged to such an extent that ancient relationships have been obscured. This finding is also supported by the haplotype sharing between many of the populations visualized in the network (Figure 3).
Fourth, despite not supporting the traditional binary subdivision of the Bedouin populations, the genetic data nevertheless provide considerable insights into their genetic history. The Ajman stood out in the Y-chromosomal analyses as outliers in the MDS plots (Figure 2) from their zero haplogroup and low haplotype diversity (Table 3) and from the population specificity of their haplotypes in the network (Figure 3). A potential explanation for these findings could be the high status of the Ajman in the Bedouin social hierarchy and lack of mixing with other populations (Dickson, 1967). To this day, the Ajman tribe is known for its zealous view of consanguineous marriage and tribal relations.
The Ajman contrast with the Aniza, who show the highest diversity among the Bedouin, with six haplogroups detected (Table 3). This observation fits several aspects of their history. Their traditional territory spanned a very wide geographical area. Their first mention in history was before the initiation of Islam, in which they were found in the Najd region of modern Saudi Arabia. From the start of Islam and through to the Ottoman era, this tribe, through conquest and raiding, extended over all of Saudi Arabia, Syria and North towards Iraqi Kurdistan and Jordan (Dickson, 1956). Slavery among the chieftains was a sign of power and wealth. Aniza tribes similar to those of Banu-Hilal and Banu Sulaym are believed to have participated in the conquest of North Africa in the 11th century CE (Abun-Nasr, 1987), possibly leading to the introduction of E-M78 chromosomes. Between the Umayyad and the Abbasid Empires, many slaves were brought from Iberia, especially Cordoba and Andalusia (Bassaam, 1977). Furthermore, envoys of the British Empire to the Persian Gulf and the Middle East between the early 19th and early 20th centuries CE mention in their memoirs the enslaving of Georgians, Armenians and Circassians (Philby, 1923). These slaves were naturalized and given tribal affiliations under slavery abolishment treaties signed between the tribal chieftains—or emirs—and the envoys of the British Empire. This could be a reason for the presence of R1b1 and G2* Y chromosomes.
The Awazim stand out because of their high frequency of haplogroup E-M123 (24.3%), the highest yet reported in any population. The highest frequency reported earlier (23.5%) was in Ethiopians from Amhara (Cruciani et al., 2004). The Awazim also showed the presence of haplogroup R2 (R-M124), characteristic of South Asia (Sengupta et al., 2006). There may be a possible link to the Roma (‘Gypsy’) migrations. It has also been proposed that the Awazim may have originated from the Caucasus—also consistent with the theory of ‘Suluba’ (Bell and Richmond, 1937). The presence of markers M123 and M124 may also suggest that slaves brought before the initiation of Islam and after the passing of the four Caliph eras (Lewis, 1990) have been naturalized within these tribes—perhaps when slavery was abolished.
The Shimar sample carried two main haplogroups—J1 (at 52.3%) and R1a1 (at 42.8%)—with a small percentage of G2 (4.76%). A historical explanation could be that the Shimar trace their origins to two regions: Iraq (Philby, 1923) and Saudi Arabia. The Shimar resided in northern Najd—currently in Saudi Arabia—after their migration during pre-Islamic times from Taye in Yemen. Their migration to the North into Iraq ended during the 17th century CE (Subahai, 1996; Khuraysi, 1998; Williamson and Basri, 1999). As they settled in Iraq, the Shimar gave up nomadism and became urbanized in towns and cities—most notably in Mosul (Khuraysi, 1998). It is said that the Shimar are now of two main antecedents—Iraqi Shimar and Saudi Shimar—and each refers to itself as ‘the Noble Tribe’ (personal communication, Shimar of Kuwait). The haplogroup R1a1-M17 is found at substantial frequency in Iraq, so limited gene flow or subsequent drift (or both) could account for its presence in the Shimar—although later migrations, slavery and trade provide alternative explanations.
In all, except for the lack of subdivision into Adnani and Qahtani lineages, the genetic data fit remarkably well with the expectations for small nomadic groups in the Arabian Peninsula, and provide independent support for many aspects of the traditional histories of these populations.
References
Abun-Nasr JM (1987). A History of the Maghrib in the Islamic Period. Cambridge University Press: Cambridge and New York.
Al-Bustan S, El-Zawahri M, Al-Azmi D, Al-Bashir AA (2002). Allele frequencies and molecular genotyping of the ABO blood group system in a Kuwaiti population. Int J Hematol 75: 147–153.
Al-Hilli F (1985). The ABO and Rh blood groups in Bahrain, Arabian Gulf. Hum Biol 57: 441–447.
Al-Nassar KE, Conneally PM, Palmer CG, Yu P (1981). The genetic structure of the Kuwaiti population. I. Distribution of 17 markers with genetic distance analysis. Hum Genet 57: 192–198.
al-Nassar KE, Mathew J, Thomas N, Fatania HR (1995). HLA-DQ alpha allele and genotype frequencies in a native Kuwaiti population. Forensic Sci Int 72: 65–69.
al-Nassar KE, Mathew J, Thomas N, Fatania HR (1996). Analysis of the D1S80 (pMCT118) VNTR locus polymorphism in a native Kuwaiti population by the polymerase chain reaction. Forensic Sci Int 78: 131–138.
Bandelt HJ, Forster P, Röhl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Bassaam AA (1977). Ulamaa Najd Khilaal Sitat Quroon. Muktabat Al-Nahdha Al-Hadeethah: Damascus.
Bell GL, Richmond EB (1937). The Earlier Letters of Gertrude Bell. Liveright Publishing Co.: New York.
Berger B, Lindinger A, Niederstatter H, Grubwieser P, Parson W (2005). Y-STR typing of an Austrian population sample using a 17-loci multiplex PCR assay. Int J Legal Med 119: 241–246.
Cadenas AM, Zhivotovsky LA, Cavalli-Sforza LL, Underhill PA, Herrera RJ (2008). Y-chromosome diversity characterizes the Gulf of Oman. Eur J Hum Genet 16: 374–386.
Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L et al. (2002). A human genome diversity cell line panel. Science 296: 261–262.
Carvalho M, Anjos MJ, Andrade L, Lopes V, Santos MV, Gamero JJ et al. (2003). Y-chromosome STR haplotypes in two population samples: Azores Islands and Central Portugal. Forensic Sci Int 134: 29–35.
Chang YM, Perumal R, Keat PY, Kuehn DL (2007). Haplotype diversity of 16 Y-chromosomal STRs in three main ethnic populations (Malays, Chinese and Indians) in Malaysia. Forensic Sci Int 167: 70–76.
Cherni L, Pereira L, Goios A, Loueslati BY, Khodjet el Khil H, Gomes I et al. (2005). Y-chromosomal STR haplotypes in three ethnic groups and one cosmopolitan population from Tunisia. Forensic Sci Int 152: 95–99.
Collins PJ, Hennessy LK, Leibelt CS, Roby RK, Reeder DJ, Foxall PA (2004). Developmental validation of a single-tube amplification of the 13 CODIS STR loci, D2S1338, D19S433, and amelogenin: the AmpFSTR® identifiler PCR amplification kit. J Forensic Sci 49: 1265–1277.
Cruciani F, La Fratta R, Santolamazza P, Sellitto D, Pascone R, Moral P et al. (2004). Phylogeographic analysis of haplohroup E3b (E-M215) Y chromosomes reveals multiple migratory events within and out of Africa. Am J Hum Genet 74: 1014–1022.
Dickson HRP (1956). Kuwait and Her Neighbours. Allen & Unwin: London.
Dickson HRP (1967). The Arab of the Desert: A Glimpse into Badawin Life in Kuwait and Sau'di Arabia. Allen & Unwin: London.
Hallenberg C, Simonsen B, Sanchez J, Morling N (2005). Y-chromosome STR haplotypes in Somalis. Forensic Sci Int 151: 317–321.
Henke J, Henke L, Chatthopadhyay P, Kayser M, Dulmer M, Cleef S et al. (2001). Application of Y-chromosomal STR haplotypes to forensic genetics. Croat Med J 42: 292–297.
Jobling MA, Hurles ME, Tyler-Smith C (2004). Human Evolutionary Genetics. Garland Science: New York and Abingdon.
Jobling MA, Tyler-Smith C (2003). The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 4: 598–612.
Khuraysi H-A (1998). Qabilat Shammar: Mutaba'ah Wa-tahlil. Dar al-Saqi: Beirut.
Lewis B (1990). Race and Slavery in the Middle East: An Historical Enquiry. Oxford University Press: New York.
Lopez AM, Alvarez S, Gusmao L, Alves C, Mesa MS, Albentosa A et al. (2004). Population data for 16 Y-chromosome STRs in four populations from Pyrenees (Spain). Forensic Sci Int 140: 125–129.
Martinez-Jarreta B, Nievas P, Abecia E, Hinojal R, Budowle B (2003). Haplotype distribution of nine Y-chromosome STR-loci in two northern Spanish populations (Asturias and Aragon). J Forensic Sci 48: 204–205.
Mulero JJ, Chang CW, Calandro LM, Green RL, Li Y, Johnson CL et al. (2006). Development and validation of the AmpFSTR® YfilerTM PCR amplification kit: a male specific, single amplification 17 Y-STR multiplex system. J Forensic Sci 51: 64–75.
Nebel A, Filon D, Brinkmann B, Majumder PP, Faerman M, Oppenheim A (2001). The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am J Hum Genet 69: 1095–1112.
Park MJ, Lee HY, Yoo JE, Chung U, Lee SY, Shin KJ (2005). Forensic evaluation and haplotypes of 19 Y-chromosomal STR loci in Koreans. Forensic Sci Int 152: 133–147.
Philby HSJB (1923). The Heart of Arabia: A Record of Travel & Exploration. G. P. Putnam and Son: New York.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Qamar R, Ayub Q, Mohyuddin A, Helgason A, Mazhar K, Mansoor A et al. (2002). Y-chromosomal DNA variation in Pakistan. Am J Hum Genet 70: 1107–1124.
Rebala K, Szczerkowska Z (2005). Polish population study on Y chromosome haplotypes defined by 18 STR loci. Int J Legal Med 119: 303–305.
Roewer L, Croucher PJ, Willuweit S, Lu TT, Kayser M, Lessig R et al. (2005). Signature of recent historical events in the European Y-chromosomal STR haplotype distribution. Hum Genet 116: 279–291.
Roewer L, Krawczak M, Willuweit S, Nagy M, Alves C, Amorim A et al. (2001). Online reference database of European Y-chromosomal short tandem repeat (STR) haplotypes. Forensic Sci Int 118: 106–113.
Schneider S, Roessli D, Excoffier L (2000). Arelquin: A Software for Population Genetics Data Analysis. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva, Geneva, Switzerland.
Sebetan IM, Hajar HA (1998). Analysis of the short tandem repeat (STR) locus HumVWA in a Qatari population. Forensic Sci Int 95: 169–171.
Sengupta S, Zhivotovsky LA, King R, Mehdi SQ, Edmonds CA, Chow CE et al. (2006). Polarity and temporality of high-resolution Y-chromosome distributions in India identify both indigenous and exogenous expansions and reveal minor genetic influence of Central Asian pastoralists. Am J Hum Genet 78: 202–221.
Singh Sawhney K, Sunderland E, Woolley V (1984). Genetic polymorphisms in the Kuwaiti Arabs. Hum Hered 34: 303–307.
Subahai A-H-A (1996). Qabilat Shammar Wa-umara'uha. Dar Ma'd lil-Tiba'ah wa-al-Nashr wa-al-Tawzi: Dimashq.
Turrina S, Atzei R, De Leo D (2006). Y-chromosomal STR haplotypes in a Northeast Italian population sample using 17plex loci PCR assay. Int J Legal Med 120: 56–59.
Williamson JF, Basri M (1999). Qabilat Shammar al-Arabiyah, Makanatuha Wa-tarikhuha al-siyasi 1800–1958. Dar al-Hikmah: London.
Wilson IJ, Weale ME, Balding DJ (2003). Inferences from DNA data: population histories, evolutionary processes and forensic match probabilities. J R Statist Soc A 166: 155–188.
Xue Y, Zerjal T, Bao W, Zhu S, Shu Q, Xu J et al. (2006). Male demography in East Asia: a north-south contrast in human population expansion times. Genetics 172: 2431–2439.
Zhivotovsky LA, Underhill PA, Cinnioglu C, Kayser M, Morar B, Kivisild T et al. (2004). The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am J Hum Genet 74: 50–61.
Acknowledgements
The authors thank all tribes and participants for their time and effort in aiding sample collection, Lutz Roewer for help with comparative data and Peter Underhill for his comments on the manuscript. TM was supported by the Amiri Diwan of Kuwait and the International Financial Advisors-Kuwait (IFA). YX and CTS were supported by The Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Rights and permissions
About this article
Cite this article
Mohammad, T., Xue, Y., Evison, M. et al. Genetic structure of nomadic Bedouin from Kuwait. Heredity 103, 425–433 (2009). https://doi.org/10.1038/hdy.2009.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.72
Keywords
This article is cited by
-
Burden of Mendelian disorders in a large Middle Eastern biobank
Genome Medicine (2024)
-
Unraveling a fine-scale high genetic heterogeneity and recent continental connections of an Arabian Peninsula population
European Journal of Human Genetics (2022)
-
Deciphering the fine-structure of tribal admixture in the Bedouin population using genomic data
Heredity (2014)
