
Abstract
We report extraordinary variation in the number and the chromosomal location of ribosomal DNA (rDNA) arrays within populations of the alpine grasshopper Podisma pedestris; even greater differences were found between populations. The sites were detected by in situ hybridisation of labelled rDNA to chromosomal preparations. The total number of rDNA sites in an individual varied from three to thirteen. In the most extreme case, individuals from populations only 10 km apart had no rDNA loci in common. A survey of the geographical distribution of this variation identified clusters of populations with relatively similar chromosomal distribution of rDNA loci. These clusters correspond to those identified earlier by analysis of rDNA sequences. To explain this geographical clustering, we reconstructed the post-glacial colonisation of the region by assuming that the species’ distribution has ascended to its current altitudinal range as the climate warmed. The reconstruction suggests that each cluster is descended from a colonisation route up a different alpine valley. That history would imply rapid establishment of rDNA differences, conceivably during the last 10 000 years since the last glaciation. The proposal for rapid change is consistent with the extensive within-population variation, which indicates that the processes responsible for the change in rDNA's chromosomal location continue to occur at a higher rate. We discuss whether our reconstruction of colonisation routes implies movement of the hybrid zone, which would indicate that a neo-XY sex chromosome system has spread through extant populations.
Similar content being viewed by others
Introduction
Ribosomal DNA (rDNA) genes code for the RNA component of ribosomes, which is essential for their role in transcription (McLain et al., 1995). Eukaryotic genomes contain multiple copies of their rDNA genes, presumably because exceptionally high quantities of RNA transcripts are required; it is not atypical for 50% of all RNA exported from the nucleus to be rRNA (Russel and Zomerdijk, 2005). Ribosomal DNA genes are organised in tandem arrays, each transcribed from a single promoter by RNA polymerase I. Each repeating unit in an array consists of an intergenic spacer involved in regulation (Reeder, 1990) then, typically, three conserved genes (18S, 5.8S and 28S) separated by more variable intergenic spacers (ITS1 and ITS2) and an external transcribed spacer (Hemleben and Zentgraf, 1994). Transcription of rDNA takes place in the nucleolar organising region, which can be shown by the cytogenetic technique of silver staining.
Ribosomal DNA sequence is extensively used for phylogenetic reconstruction for two reasons. One is that some parts of rDNA are among the most conserved sequences known (Eickbush and Eickbush, 2007), so homologues can be readily identified. Second, the different units within any one species tend to be homogenous (especially members of the same array) (Liao, 1999) a pattern termed concerted evolution (Zimmer et al., 1980). The homogenising processes that maintain the similarity between units within a species probably include unequal crossing-over and gene conversion (Eickbush and Eickbush, 2007), although the relative importance of each mechanism is not clear and their contribution may vary depending on the genome in question.
In contrast to this conventional picture, there have been recent reports of species that do not exhibit the high within-species rDNA homogeneity characteristic of concerted evolution. They include aphids (Fenton et al., 1998), flatworms (Carranza et al., 1999), mosquitoes (Li and Wilkerson, 2007), trypanosomes (Beltrame-Botelho et al., 2005), the phylum Apicomplexa (Rooney, 2004) and actinomycetous bacteria (Wang et al., 1997). We have recently described the case of extensive within-individual and between-individual variability in ITS1 rDNA sequences of the grasshopper Podisma pedestris (Keller et al., 2006, 2008). Individuals from the same, or neighbouring, populations had more similar sequences, suggesting that ancestral polymorphisms can persist for extended periods of time. However, bursts of rapid change in the frequency of certain rDNA variants also seem to have occurred, as several individuals had an rDNA sequence composition that differed markedly from their population, or any other (Keller et al., 2008).
Occasionally, studies have detected changes in the chromosomal location and size of specific rDNA arrays (loci). For example, the location of arrays differs between sibling species within the Drosophila melanogaster complex (Lohe and Roberts, 1990). More rapid change is suggested by differences between populations of the brown trout, Salmo trutta (Castro et al., 2001), and the grasshopper, Eyprepocnemis plorans (Cabrero et al., 2003). In D. melanogaster, the number of rDNA units on the X chromosome varied more than twofold between different lines, after 400 generations of divergence in the laboratory (Averbeck and Eickbush, 2005).
The variation in the dynamics of rDNA evolution between different species is puzzling, as the underlying molecular biology is presumed to be the same. One possibility is that general properties of the genome are relevant. The genomic environment of P. pedestris is quite different from a typical model organism. It contains 16.93 pg of DNA (Westerman et al., 1987), roughly 100 times more than the genome of D. melanogaster. It includes multiple copies of mitochondrial pseudogenes in the nuclear DNA (Numts). Analysis of Numts suggests that point mutations are more frequent than deletions in the P. pedestris nuclear genome, the inverse situation to that in D. melanogaster (Bensasson et al., 2001). By studying such atypical genomes we hope to understand how the internal genomic environment can affect its evolution.
Scientific interest in P. pedestris initially arose because it is subdivided into chromosomal races. It is a flightless grasshopper adapted to steppe-like climate. It is widely distributed in Northern Eurasia (Klochanova, 1953; Latchininsky, 1997). In the French Alps, the area of study, the species’ range has retreated to higher altitudes in the last 10 000 years, because of post-glacial climate warming (Hewitt, 1999). Abundant populations are typically found between 1500 and 2500 m (Nichols and Hewitt, 1986). The sex chromosome system is XO for males and XX for females throughout most of the range of the species (the unfused race). In the Southern Alps, a Robertsonian fusion between the L3 autosome and the X chromosome has become established to produce a neo-XY system, with XY males and XX females (the fused race) (John and Hewitt, 1970; Hewitt, 1975).
In places where the two chromosomal races meet, a narrow cline in the frequency of the fusion is formed, which is 800 m wide (Barton and Hewitt, 1981b). The hybrid zone does not seem to coincide with an environmental gradient (Barton and Hewitt, 1981a; Nichols and Hewitt, 1988), indicating that it is a tension zone maintained by some form of selection against introgressing genotypes. This hypothesis is supported by crossing experiments, which detected inviability (Barton and Hewitt, 1981b). Before our rDNA sequence analysis, few fixed differences had been found between the races: one involved a restriction enzyme variant in rDNA (Dallas et al., 1988) and another was a 5% reduction in total genomic DNA (Westerman et al., 1987), which could conceivably involve rDNA. Both differences remained in strong linkage disequilibrium with the fusion in one well studied transect across the hybrid zone. No obvious size difference between the chromosomes of the two races has been discerned by light microscopy and no other clear differentiation between the races has been found in morphology or allozymes (Halliday et al., 1983; Nichols and Hewitt, 1986).
In this paper, we continue the investigation of the rDNA variation within P. pedestris by identifying the chromosomal location (loci) of the rDNA tandem arrays. We employ fluorescent in situ hybridisation (FISH) with an rDNA probe, to detect chromosomal ‘sites’, where there are rDNA arrays. We exploit a subset of the samples used by Keller et al. (2008) so that direct comparisons can be drawn with their results.
Materials and methods
Sample collection and preparation
Details of the sampling are provided in Keller et al. (2008). Coordinates are listed in the Supplementary information. Briefly, two areas of the Southern French Alps (one close to the village of Seyne and the other close to Casterino) were sampled, usually between 1500 and 2000 m, where P. pedestris is most prevalent and accessible. Male grasshoppers were collected within a 20 m radius for each population and dissected in the field. The testes were fixed in freshly prepared 3:1 ethanol:acetic acid solution, the fixative was replaced with ethanol and the samples were kept at 4 °C for long-term storage.
FISH technique and protocols
The FISH procedures were based on Schwarzacher and Heslop-Harrison (2000).
Probe information.
Ribosomal DNA probe
The rDNA probe was either a labelled pTa71 clone (Gerlach and Bedbrook, 1979) (a widely used standard rDNA probe), or a 35S PCR product amplified from P. pedestris 18S DNA. Preliminary two-colour FISH experiments using both probes simultaneously indicated that they resulted in the same labelling profile. This result indicates that the probes are detecting rDNA rather than non-specific hybridisation with repetitive sequences. The 35S PCR used the primers 18S_F (5′-ACGTTACTTGGATAACTGTGGT-3′) and ITS2_B (5′-TATGCTTAAATTCAGGGGGT-3′) (Beebe and Saul, 1995), which amplify a 2.5 kb region that includes most of the 18S gene and extends until the very beginning of the 28S gene. PCR amplifications were carried out using a PTC-220 DNA Engine DyadTM Peltier Thermal Cycler (MJ Research, Essex, UK). Each reaction (25 μl) contained approximately 5–10 ng of genomic DNA, 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.5 pmol of each primer, 0.5 units of BIOTAQ DNA Polymerase (Bioline, London, UK) and 1 × NH4+ buffer. An initial denaturation step of 3 min at 94 °C was followed by 31 cycles of 30 s at 94 °C, 30 s at 52 °C, 3 min at 72 °C and a final elongation step of 10 min at 72 °C.
Telomeric probe: A telomeric probe was also used in the FISH survey, primarily as a marker to help interpret the configuration of the bivalents at meiosis, and hence to distinguish the chromosomes. The sequence of the telomeric repeat of all Orthoptera studied so far is (TTAGG)n (López-Fernández et al., 2006). The telomeric probe was generated by PCR reaction using the primers Telo_3 (5′-TAGGTTAGGTTAGGTTAGGT-3′) and Telo_4 (5′-CTAACCTAACCTAACCTAAC-3′) alone. They hybridize with each other and hence produce a product in the absence of other template DNA (Sahara et al., 1999). The PCR reaction followed the protocol in Ijdo et al. (1991). PCR amplifications were carried out using a PTC-220 DNA Engine DyadTM Peltier Thermal Cycler (MJ Research). Each reaction (100 μl) contained 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.1 pmol of each primer, 2 units of BIOTAQ DNA Polymerase (Bioline) and 1 × NH4+ buffer. An initial denaturation step of 3 min at 94 °C was followed by 10 cycles of 60 s at 94 °C, 30 s at 52 °C, 60 s at 72 °C and 30 cycles of 60 s at 94 °C, 30 s at 52 °C, 90 s at 72 °C and a final elongation step of 5 min at 72 °C.
Nick translation
Probe labelling was performed by nick translation following the protocol provided in Schwarzacher and Heslop-Harrison (2000). Each reaction (50 μl) contained nick translation buffer (0.05 M Tris-HCl, pH 7.8, 0.005 M MgCl2, 0.05 mg ml−1 bovine serum albumin, nuclease-free water (Sigma-Aldrich, Dorset, UK), 0.2 mM dithiothreitol, 0.005 mM of each dCTP, dGTP and dATP, and 2.5 units of DNA polymerase I and 0.002 units of DNase I (Invitrogen, Paisley, UK). For biotin labelling, 2.5 μl of 0.4 mM biotin-11-dUTP (Sigma, Dorset, UK), made up from powder in 100 mM Tris-HCl pH 7.5, were added to the reaction. For digoxigenin labelling, 1 μl of 0.35 mM digoxigenin-11-dUTP (Roche Biochemicals, Burgess Hill, UK) and 0.65 mM dTTP were added to the reaction. The product was stored at −20 °C until required, and could be used without further purification. The total DNA produced per reaction was judged by eye on an agarose gel and standardised to ∼1 μg.
Chromosome preparations
Meiotic chromosome squashes were prepared by squashing the fixed testes in 50–75% acetic acid on polylysine coated slides (VWR, Lutterworth, UK). The slides were frozen on a column of dry ice for 30 min and the coverslip was flicked off using a scalpel blade. The slides were air-dried and stored at −20 °C, typically for 2 weeks, before going through the FISH protocol.
In situ hybridisation
Typically, 12 slides were treated at a time. Washes were made in Coplin jars (six slides each). The slides were treated with 100 μg ml−1 RNase A (Sigma, Dorset, UK) in Tris-HCl for 1 h at 37 °C in a humid chamber. They were then washed with 2 × saline-sodium citrate (SSC) (0.3 M NaCl, 0.03 M sodium citrate) and treated with 0.25 μg ml−1 pepsin (Sigma-Aldrich) in 0.01 M HCl for 5–15 min, depending on the amount of cytoplasm remaining around the meiotic chromosomes. After washing with 2 × SSC, the slides were fixed in freshly depolymerised paraformaldehyde 0.025% (w/v) at room temperature. They were then washed in 2 × SSC and dried in an ethanol series.
Next, 0.5–1.2 μl of biotin- or digoxigenin-labelled DNA (0.1 μg ml−1) was added to the probe solution (50% formamide, 0.1% dextran sulphate, 0.01 × SSC, 0.0375% (w/v) sodium dodecyl sulphate). The probe mix was denatured at 76 °C for 10 min before hybridisation and kept on ice. It was applied to the slides directly and hybridisation was performed on the hot plates of a PTC-220 DNA Engine DyadTM Peltier Thermal Cycler (MJ Research) using the following program: 72 °C for 7 min, 55 °C for 2 min, 50 °C for 30 s, 45 °C for 1 min, 42 °C for 2 min, 40 °C for 5 min, 38 °C for 5 min, 37 °C thereafter (until the product was transferred to the humid chamber, typically after 1 h). The slides were incubated overnight at 37 °C in a humid chamber.
Post-hybridisation washing
The slides were washed in the following: 2 × SSC for 2 min at room temperature, a stringent wash (20% formamide in 0.1 × SSC) at 42 °C for 10 min, 2 × SSC at 42 °C for at least 10 min and then washed at room temperature in 4 × SSC Tween (0.6 M NaCl, 0.06 M sodium citrate, 0.002% v/v Tween). Sites of hybridisation of the digoxigenin- and biotin-labelled probes were detected simultaneously (two-colour FISH) using 10 μg ml−1 antidioxigenin-FITC (Roche Biochemicals) and 5 μg ml−1 avidin-Cy3 (Amersham Pharmacia, Buckinghamshire, UK), which were added to the slide and incubated at 37 °C for 1 h. A drop of Vectashield including DAPI (Vector laboratories, Peterborough, UK) was added on each slide to counterstain the chromatin and to stabilise the fluorochromes.
Imaging
Fluorescence signal was viewed with a Leica DMRA2 microscope, captured with a Hamanabu ORCA-ER digital camera and processed with the software Openlab V3.15 (Improvision Ltd, Coventry, UK) on a Power Macintosh computer. Images were exported in Tiff format (publication quality) to Adobe Photoshop (Adobe Systems, Uxbridge, UK) for further manipulation. The images were treated uniformly by changing contrast, brightness and colour balance only. Only individuals that provided at least three images from different cells, each with clear probe localisation, were included in the results.
Inferring colonisation routes
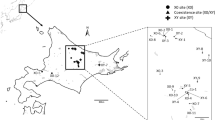
The past distribution of P. pedestris populations was assessed by starting from a map of the 1000 m altitudinal range (between 1500 and 2500 m, Figure 2d), in which most P. pedestris populations are currently found in the region. This band was displaced downwards to represent colder climates (Figures 2b and c). The assumption is that, within this range, there would have been suitable habitat, which would have been colonised rapidly compared with the rate of climate change. The actual range would have been a subset of the area indicated, because of other ecological limitations. Nevertheless, the reconstruction is sufficient to suggest (a) the possible colonisation routes and (b) the subdivision of the range.

Comparison of the FISH and sequencing data. Panel (a) is a principal component plot based on the frequency of rDNA variants within each individual (redrawn from Keller et al., 2008). Each point represents an individual grasshopper (triangles, unfused; squares, fused). Filled shapes represent samples for which the new FISH data are also available; their colours indicate the cytogenetic group from Table 1. Circled individuals (in panel a) were collected at the site marked by an asterisk on the maps (Colle della Maddalena), see Discussion. The pie charts on the maps show the proportion of each cytogenetic group. The light grey area represents the habitable altitudinal range. The upper altitudinal limit of the light grey area in each of (b–d) is given in its top right corner. Panel d shows the current course of the hybrid zone (as determined by Hewitt, 1975) and the towns of Seyne and Casterino (marked S and C). The numbered arrows indicate valleys, which may have been routes of colonisation.
Altitude and GPS data were manipulated in ArcViewGIS v9.2. The elevation data for the map were kindly provided by Patrick Coquillard. The GPS data for the years 2003–2005 were estimated from maps whereas the 2006 sampling locations were obtained directly in the field by a GPS device.
Results
Extreme rDNA locus variability
There was great variation in both the number and location of chromosomal sites detected by the rDNA probe. We found differences between populations, and often between individuals within the same population. We shall use the term ‘sites’ for a chromosomal region where signal was detected; hence at any one locus there could be none, one or two sites. If there was only one site, the locus was classified as monosomic. When calculating the total number of rDNA sites in an individual, a homologous pair of sites was counted as two observations irrespective of any difference in size, whereas a monosomic locus was counted as one. The FISH method may not detect small numbers of rDNA units, hence sites with small numbers of units cannot be distinguished from those with none.
The rDNA sites were categorised as being on large, medium or small bivalents, following John and Hewitt (1970) and as being proximal (near to the centromere), or distal (close to the far chromosome end; see Figure 1). All P. pedestris chromosomes seem to be telocentric, with the exception of the fused X chromosome. Details of the FISH data are provided in Supplementary file S1. Individuals with as few as three and as many as 13 rDNA sites were found. Surprisingly, monosomic rDNA loci were in the majority in some populations. Consequently, the maximum possible number of sites may be greater than the observed maximum of 13 rDNA sites.

Ideograms of a karyotype from each of the cytogenetic groups (original images are in Supplementary Figure S2). Black stars show rDNA sites that are present in most members of a group. White stars indicate rDNA sites that are absent in some members of the group. The darker chromosome arm is the ancestral X chromosome. Note that because the chromosomes within each size category (small, medium and large) cannot be distinguished, loci can only be provisionally classified as consistently present or not. The second letter of each cytogenetic group code indicates the nature of the X chromosome: F=fused; U=unfused.
Division of the populations into cytogenetic groups
Some rDNA loci were consistently present in almost all individuals from a sampled population. These loci are indicated in bold in Table 1 and were used to divide the populations into geographically contiguous groups. First, the populations were divided into fused and unfused as indicated by the second letter of the cytogenetic group name (F or U in Table 1). The fused populations were further subdivided into the Casterino fused (CF) group, which had a proximal rDNA site on the X chromosome, and the Seyne fused group, which did not. The Mélèzes unfused populations had no distal rDNA sites on the X chromosome (and very few on autosomes). The remaining unfused populations could be subdivided on the basis of the median number of distal autosomal rDNA sites, two in the case of Lac Noir unfused group and four for Casterino unfused group. Figure 1 illustrates an individual from each cytogenetic group. FISH images from the same individuals are provided in Supplementary File S2.
The cytogenetic groupings are compared with those of Keller et al. (2008) in Figure 2. Shown in Figure 2a is a principal component analysis plot based on SNP variants in rDNA, in which each symbol represents an individual. The genetic clusters are distinguished by colour and the same colours were used to show the geographical distribution of each group on the adjoining maps (Figures 2b–d). With one exception, the groupings obtained from the new cytogenetic data correspond to those identified earlier from the molecular genetic data. The one exception is a population (Colle della Maddalena) geographically isolated from the rest (see Discussion).
Hybridisation of the telomere specific (TTAGG)n probe was observed on the centromeres of all fused X chromosomes. It is conceivable that telomeric DNA was left at this location after the fusion between two telocentric chromosomes (the ancestral X chromosome and an L3 autosome) and has subsequently been retained.
Discussion
Extensive variation in number and location of rDNA sites
We have found extensive variation in the number and location of rDNA sites both between and within cytogenetic groups of P. pedestris (Table 1). A few comparable reports exist from a wide range of taxa, including beetles of the genus Zabrus (Sánchez-Gea et al., 2000), the red abalone Haliotis rufescens (Gallardo-Escárate et al., 2005), the fish Astyanax scabripinnis (Mantovani et al., 2005), the grasshopper E. plorans (Cabrero et al., 2003) and onions Allium spp. (Schubert and Wobus, 1985). P. pedestris stands out for two reasons. One is the extent of variation in the number of rDNA sites and of monosomicity (the occurrence of loci at which rDNA occurs on only one of the two homologous chromosomes), especially in the Mélèzes unfused populations, where there were from five to thirteen sites, of which 0–75% were monosomic. Second, variation in rDNA loci (in number, location and monosomicity) was extremely localised geographically. The Casterino area displayed little variation from population to population, and limited polymorphism within cytogenetic groups. By contrast, the most extreme example of localised variation is provided by the Mélèzes unfused and Seyne fused cytogenetic groups. They occur only 10 km apart, yet include individuals with no rDNA loci in common.
Just as high levels of heterozygosity would be observed at a conventional locus with a high-mutation rate, the high levels of rDNA locus monosomicity in P. pedestris could be explained by high rates of origin and loss of rDNA arrays. As these changes have persisted, they do not seem to be strongly deleterious, perhaps, because they involve non-expressed rDNA. Indeed, Keller et al. (2006) estimated that 75% (range 71–78%) of the total rDNA is methylated or has atypical DNA sequence, using Southern blot experiments. Hence, it is conceivable that whole rDNA loci are composed of constitutively inactive repeats or pseudogenes.
We conjecture that the consistently present loci are likely to be the most active (that is, transcribed to produce the RNA component of ribosomes) because of selection against their removal. This interpretation corresponds with earlier published results, based on silver staining, showing that the (consistently present) distal locus of the X chromosome is active. The results were more equivocal for the autosomes and not readily interpretable (Gosálvez et al., 1988, J Gosálvez, C Lopéz-Fernández and JL Bella, personal communication). However, rDNA mobility is not confined to inactive loci: an evolutionary perspective allows us to conclude that active rDNA must have moved in some cases. In particular, active sites must have moved as the Mélèzes unfused and Seyne fused groups diverged, because they have no rDNA loci in common.
Recolonisation after the last glacial maximum
The subdivision of the study populations into different groups using the cytogenetic data reported here (number and location of rDNA sites) coincides with the subdivision based on rDNA sequence variation (Figure 2). This genetic structure is probably the result of repeated extinction and recolonisation events. The area was recently glaciated (Hewitt, 1999) and before that, there were multiple climatic cycles, which would have caused population expansion and contraction (every 100 000 years (Hays et al., 1976)). The geographical distribution of the cytogenetic groups can be explained by different colonisation routes along which the grasshoppers expanded into their current range after the last glacial maximum (arrows in Figures 2b and c). As P. pedestris rDNA harbours extensive within-individual and within-population diversity, the allele frequencies could diverge by genetic drift. Range expansion can cause rapid genetic drift as populations are founded and grow at the expanding margin of a species range (Nichols and Hewitt, 1994; Ibrahim et al., 1996; Excoffier and Ray, 2008). The cytogenetic and molecular differences could both have become established during the same expansions, which would explain why they correspond. The only minor discrepancy is a population to the northeast, Colle della Maddalena (marked by asterisk in Figure 2). Its rDNA sequence composition differs from Casterino unfused group (its cytogenetic group) in being fixed for six polymorphic sites that are common in other unfused populations. Perhaps, it accumulated differences from other Casterino unfused populations during an expansion from the Italian side of the border.
The chromosomal fusion may also have become established through genetic drift potentially during population expansion from a glacial refugium. The cytogenetic and molecular differences between the different fused populations could then have accumulated over a number of cycles. Attributing the current distribution of the fused race exclusively to extinction and recolonisation processes is not entirely straightforward, however, the current position of the hybrid zone does not seem to be at the junction of two expansions. If the expansion marked (i) in Figure 2b was comprised of unfused individuals it would seem to be inevitable that the route marked (ii) would also have been unfused; yet, fused populations are currently observed further up that valley. It is possible to propose alternative explanations; for example, a Northern unfused refuge in valley (i) from which the unfused regions were populated whereas the fused race arrived from the South (iii) from where it could also have reached the Casterino area and the other populations known to be fused (routes (iv)).
An alternative explanation is that the fused chromosomes spread northwards once unfused populations had become established. Conventional hybrid zone theory suggests the zone would not move, once formed (Hewitt, 1988), however, evidence that some zones do actually move is accumulating (reviewed in Buggs, 2007) and recent theoretical developments suggest that the spread of an X-autosome fusion could be explained by the sexually antagonistic selection expected in early sex chromosome evolution (Veltsos et al., 2008). The reduced recombination between the centromeric region of the neo-Y and the fused X chromosomes (John and Hewitt, 1970) would facilitate the accumulation of alleles with sexually antagonistic effects (Charlesworth, 1996). There is some preliminary evidence consistent with the spread of the fusion into an unfused genetic background at Casterino: individuals from the hybrid zone grouped with unfused individuals based on rDNA sequence variation (Keller et al. (2008) Supplementary material).
Most interesting is the distinction between the groups Mélèzes unfused and Lac Noir unfused for which the reconstruction suggests expansion along different valleys ((v) for Mélèzes unfused and the large valley to the north of (vi) for Lac Noir unfused group; Figure 2c) from the same south-western refugium. If these populations are not derived from the same ancestral population, they must have been closely adjacent. That logic would imply that the dramatic differences between Mélèzes unfused and Lac Noir unfused group either arose during the ascent (in the last 10 000 years) or at least some time in the last climatic cycle, which in turn suggests that rDNA is particularly mobile.
Possible molecular mechanisms causing rapid change
Why should rDNA be mobile, compared with the rest of the genome? Repeated DNA motifs in rDNA units may produce sequence matches to other genomic regions and could promote non-homologous recombination (Castro et al., 2001). Such sequence similarities may be particularly frequent in P. pedestris, whose large genome (Westerman et al., 1987) will include a high proportion of repeated elements and other rapidly evolving sequences, as exemplified by mtDNA pseudogenes (Vaughan et al., 1999; Bensasson et al., 2000, 2001). For example, the transposition of mobile elements (Sánchez-Gea et al., 2000) present in rDNA to new locations could produce matches around the genome. However, phylogenetic analysis of rDNA loci location in 49 grasshopper species argues against this type of explanation; Cabrero and Camacho (2008) argue that a recombination-mediated mechanism would produce greater variation among distal loci than proximal loci, whereas they observe the opposite.
Movement of sequences to novel chromosomal locations most likely by some form of transposition, followed by rapid expansion and contraction of rDNA arrays (Dubcovsky and Dvorak, 1995), could produce the variation observed in P. pedestris. Unequal crossing-over (because of mis-alignment of homologous chromosomes in regions of tandem repeats), which has been implicated in concerted evolution of rDNA, would also cause changes in the number of rDNA units within arrays (Eickbush and Eickbush, 2007). However, it is unclear whether the rate of such changes would be sufficient to explain the variation observed in P. pedestris.
Evidence of additional processes that rapidly increase the number of rDNA units has been obtained from model organisms (Saccharomyces cerevisiae, D. melanogaster). A mechanism may be triggered when the number of rDNA genes falls sufficiently to impair fitness (Procunier and Tartof, 1978; Kobayashi et al., 1998; Kobayashi and Ganley, 2005). As well as low-copy number, DNA damage might initiate some form of repair mechanism; Russel and Zomerdijk (2005) have speculated that, as the rDNA polymerase (pol I) is so consistently active in the cell cycle, it might be under close molecular surveillance for stalled transcription. Damage to rDNA might therefore be particularly prone to set off a repair cascade. Whatever the proximate trigger, different genetic elements have been identified which seem to facilitate rapid amplification either in cis (Borisjuk et al., 2000) or in trans (Lohe and Roberts, 1990). This sort of expansion could potentially lead to very rapid changes in rDNA sequence composition within populations. In fact, we have earlier found rare individuals with completely different frequency of rDNA sequence variants from the rest of their cytogenetic group, yet, with no differences in their rDNA loci (Keller et al., 2008). As yet, the molecular mechanisms responsible for any of these observations remain unknown.
References
Averbeck KT, Eickbush TH (2005). Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171: 1837–1846.
Barton NH, Hewitt GM (1981a). A chromosomal cline in the grasshopper Podisma pedestris. Evolution 35: 1008–1018.
Barton NH, Hewitt GM (1981b). The genetic basis of hybrid inviability in the grasshopper Podisma pedestris. Heredity 47: 367–383.
Beebe NW, Saul A (1995). Discrimination of all members of the Anopheles punctulatus complex by polymerase chain reaction-restriction fragment length polymorphism analysis. Am J Trop Med Hyg 53: 478–481.
Beltrame-Botelho IT, Gaspar-Silva D, Steindel M, Davila AMR, Grisard EC (2005). Internal transcribed spacers (its) of Trypanosoma rangeli ribosomal DNA (rDNA): a useful marker for inter-specific differentiation. Infect Genet Evol 5: 17–28.
Bensasson D, Petrov DA, Zhang D, Hartl DL, Hewitt GM (2001). Genomic gigantism: DNA loss is slow in mountain grasshoppers. Mol Biol Evol 18: 246–253.
Bensasson D, Zhang D, Hewitt GM (2000). Frequent assimilation of mitochondrial DNA by grasshopper nuclear genomes. Mol Biol Evol 17: 406–415.
Borisjuk N, Borisjuk L, Komarnytsky S, Timeva S, Hemleben V, Gleba Y et al. (2000). Tobacco ribosomal DNA spacer element stimulates amplification and expression of heterologous genes. Nat Biotechnol 18: 1303–1306.
Buggs RJA (2007). Empirical study of hybrid zone movement. Heredity 99: 301–312.
Cabrero J, Camacho JP (2008). Location and expression of ribosomal RNA genes in grasshoppers: abundance of silent and cryptic loci. Chromosome Res 16: 595–607.
Cabrero J, Perfectii F, Gómez R, Camacho JPM, Lopéz-Leon MD (2003). Population variation in the A chromosome distribution of satellite DNA and ribosomal DNA in the grasshopper Euprepocnemis plorans. Chromosome Res 11: 375–381.
Carranza S, Baguna J, Riutort M (1999). Origin and evolution of paralogous rRNA gene clusters within the flatworm family Dugesiidae (Platyhelminthes, Tricladida). J Mol Evol 49: 250–259.
Castro J, Rodriguez S, Pardo B, Sanchez L, Martinez P (2001). Population analysis of an unusual NOR-site polymorphism in brown trout (Salmo trutta L.). Heredity 86 (Part 3): 291–302.
Charlesworth B (1996). The evolution of chromosomal sex determination and dosage compensation. Curr Biol 6: 149–162.
Dallas JF, Barton NH, Dover GA (1988). Interracial rDNA variation in the grasshopper Podisma pedestris. Mol Biol Evol 5: 660–674.
Dubcovsky J, Dvorak J (1995). Ribosomal RNA multigene loci: nomads of the Triticeae genomes. Genetics 140: 1367–1377.
Eickbush TH, Eickbush DG (2007). Finely orchestrated movements: evolution of the ribosomal RNA genes. Genetics 175: 477–485.
Excoffier L, Ray N (2008). Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol Evol 23: 347–351.
Fenton B, Malloch G, Germa F (1998). A study of variation in rDNA ITS regions shows that two haplotypes coexist within a single aphid genome. Genome 41: 337–345.
Gallardo-Escárate C, Álvarez-Borrego J, Del Río-Portilla MA, Cross I, Merlo A, Rebordinos L (2005). Fluorescence in situ hybridisation of rDNA, telomeric (TTAGGG)n and (GATA)n repeats in the red abalone Haliotis rufescens (Archaeogastropoda: Haliotidae). Hereditas 142: 73–79.
Gerlach W, Bedbrook J (1979). Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res 7: 1869–1885.
Gosálvez J, Bella JL, Hewitt GM (1988). Chromosomal differentiation in Podisma pedestris: a third race. Heredity 61: 149–157.
Halliday RB, Barton NH, Hewitt GM (1983). Electrophoretic analysis of a chromosomal hybrid zone in the grasshopper Podisma pedestris. Biol J Linn Soc 19: 51–62.
Hays JD, Imbrie J, Shackleton N (1976). Variations in the Earth's orbit: pacemaker of the ice ages. Science 194: 1121–1132.
Hemleben V, Zentgraf U (1994). ‘Structural organization and regulation of transcription by RNA polymerase I of plant nuclear ribosomal genes.’ In: Novel L (ed). Results and Problems in Cell Differentiation 20: Plant Promoters and Transcription Factors. Springer-Verlag: Berlin. pp 3–24.
Hewitt GM (1975). A sex chromosome hybrid zone in the grasshopper Podisma pedestris (Orthoptera: Acrididae). Heredity 35: 375–387.
Hewitt GM (1988). Hybrid zones—natural laboratories for evolutionary studies. Trends Ecol Evol 3: 158–167.
Hewitt GM (1999). Post-glacial re-colonization of European biota. Biol J Linn Soc 68: 87–112.
Ibrahim KM, Nichols RA, Hewitt GM (1996). Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77: 282–291.
Ijdo JW, Wells RA, Baldini A, Reeders ST (1991). Improved telomere detection using a telomere repeat probe (TTAGGG)n generated by PCR. Nucleic Acids Res 19: 4780.
John B, Hewitt GM (1970). Inter-population sex chromosome polymorphism in the grasshopper Podisma pedestris. Chromosoma 31: 291–308.
Keller I, Chintauan-Marquier IC, Veltsos P, Nichols RA (2006). Ribosomal DNA in the grasshopper Podisma pedestris: Escape from concerted evolution. Genetics 174: 863–874.
Keller I, Veltsos P, Nichols RA (2008). The frequency of rDNA variants within individuals provides evidence of population history and gene flow across a grasshopper hybrid zone. Evolution 62: 833–844.
Klochanova VA (1953). The wingless grasshopper (Podisma pedestris) as a pest of forest nurseries in the steppe zone. Rev Entomol USSR 33: 78–79.
Kobayashi T, Ganley A (2005). Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309: 1581–1584.
Kobayashi T, Heck D, Nomura M, Horiuchi T (1998). Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev 12: 3821–3830.
Latchininsky AV (1997). ‘Grasshopper control in Siberia: strategies and perspectives’. In: Krall S, Pevelling R, Ba Diallo D (eds). New Strategies in Locust Control. Birkhauser: Verlag Basel, Switzerland. pp 493–502.
Li C, Wilkerson RC (2007). Intragenomic rDNA ITS2 variation in the neotropical Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culicidae). J Hered 98: 51–59.
Liao D (1999). Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet 64: 24–30.
Lohe AR, Roberts PA (1990). An unusual Y chromosome of Drosophila simulans carrying amplified rDNA spacer without rRNA genes. Genetics 125: 399–406.
López-Fernández C, Arroyo F, Fernández J, Gosálvez J (2006). Interstitial telomeric sequence blocks in constitutive pericentromeric heterochromatin from Pyrgomorpha conica (Orthoptera) are enriched in constitutive alkali-labile sites. Mutat Res 599: 36–44.
Mantovani M, Dos Santos LD, Moreira-Filho O (2005). Conserved 5S and variable 45S rDNA chromosomal localisation revealed by FISH in Astyanax scabripinnis (Pisces, Characidae). Genetica 123: 211–216.
McLain D, Wesson D, Collins F, Oliver JJ (1995). Evolution of the rDNA spacer, ITS2, in the ticks Ixodes scapularis and I. pacificus (Acari: Ixodidae). Heredity 75: 303–319.
Nichols RA, Hewitt GM (1988). Genetical and ecological differentiation across a hybrid zone. Ecol Entomol 13: 39–49.
Nichols RA, Hewitt GM (1986). Population structure and the shape of a chromosomal cline between two races of Podisma pedestris (Orthoptera: Acrididae). Biol J Linn Soc 29: 301–316.
Nichols RA, Hewitt GM (1994). The genetic consequences of long distance dispersal during colonization. Heredity 72: 312–317.
Procunier JD, Tartof KD (1978). A genetic locus having trans and contiguous cis functions that control the disproportionate replication of ribosomal RNA genes in Drosophila melanogaster. Genetics 88: 67–79.
Reeder RH (1990). rDNA synthesis in the nucleolus. Trends Genet 6: 390–394.
Rooney AP (2004). Mechanisms underlying the evolution and maintenance of functionally heterogeneous 18 s rRNA genes in apicomplexans. Mol Biol Evol 21: 1704–1711.
Russel J, Zomerdijk JCBM (2005). RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci 30: 87–96.
Sahara K, Marec F, Traut W (1999). TTAGG telomeric repeats in chromosomes of some insects and other arthropods. Chromosome Res 7: 449–460.
Sánchez-Gea JF, Serrano J, Gálian J (2000). Variability in rDNA loci in Iberian species of the genus Zabrus (Coleoptera: Carabidae) detected by fluorescence in situ hybridisation. Genome 43: 22–28.
Schubert I, Wobus U (1985). In situ hybridisation confirms jumping nucleolus organizing regions in Allium. Chromosoma 92: 143–148.
Schwarzacher T, Heslop-Harrison P (2000). Practical In Situ Hybridization. BIOS Scientific Publishers Limited: Oxford.
Vaughan HE, Heslop-Harrison JS, Hewitt GM (1999). The localisation of mitochondrial sequences to chromosomal DNA in Orthopterans. Genome 42: 874–880.
Veltsos P, Keller I, Nichols RA (2008). The inexorable spread of a newly arisen Neo-Y chromosome. PLoS Genet 4: e1000082.
Wang Y, Zhang Z, Ramanan N (1997). The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16 s rRNA genes. J Bacteriol 179: 3270–3276.
Westerman M, Barton NH, Hewitt GM (1987). Differences in DNA content between two chromosomal races of the grasshopper Podisma pedestris. Heredity 58: 221–228.
Zimmer EA, Martin S, Beverley S, Kan YW, Wilson A (1980). Rapid duplication and loss of genes coding for the alpha chains of hemoglobin. Proc Natl Acad Sci USA 77: 2158–2162.
Acknowledgements
We are grateful to Patrick Coquillard (University of Nice-Sophia-Antipolis, Parc Valrose, 06108 Nice cedex 2, France) for providing the GIS data and Andrew Leitch, Dave Horne and Roger Butlin for helpful comments and discussions. Three referees also provided valuable comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Rights and permissions
About this article
Cite this article
Veltsos, P., Keller, I. & Nichols, R. Geographically localised bursts of ribosomal DNA mobility in the grasshopper Podisma pedestris. Heredity 103, 54–61 (2009). https://doi.org/10.1038/hdy.2009.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.32
Keywords
This article is cited by
-
Cytogenetic markers reveal a reinforcement of variation in the tension zone between chromosome races in the brachypterous grasshopper Podisma sapporensis Shir. on Hokkaido Island
Scientific Reports (2019)
-
How dynamic could be the 45S rDNA cistron? An intriguing variability in a grasshopper species revealed by integration of chromosomal and genomic data
Chromosoma (2019)
-
Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids
BMC Genomics (2012)
-
Chromosomal dynamics of nucleolar organizer regions (NORs) in the house mouse: micro-evolutionary insights
Heredity (2012)
-
The linked units of 5S rDNA and U1 snDNA of razor shells (Mollusca: Bivalvia: Pharidae)
Heredity (2011)

{kind=link}