
Abstract
In the era of post-genomic research two new disciplines, Systems and Synthetic biology, act in a complementary way to shed light on the ever-increasing amount of data produced by novel high-throughput techniques. Systems biology aims at developing a formal understanding of biological processes through the development of quantitative mathematical models (bottom-up approach) and of ‘reverse engineering’ (top-down approach), whose aim is to infer the interactions among genes and proteins from experimental observations (gene regulatory networks). Synthetic biology on the other hand uses mathematical models to design novel biological ‘circuits’ (synthetic networks) able to perform specific tasks (for example, periodic expression of a gene of interest), or able to change the behavior of a biological process in a desired way (for example, modify metabolism to produce a specific compound of interest). The use of a pioneering approach that combines biology and engineering, to describe and/or invent new behaviors, could represent a valuable resource for studying complex diseases and design novel therapies. The identification of regulatory networks will help in identifying hundreds of genes that are responsible for most genetic diseases and that could serve as a starting point for therapeutic intervention. Here we present some of the main genetics and medical applications of these two emerging fields.
Similar content being viewed by others

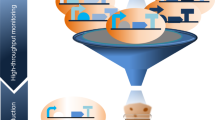

Introduction
One of the main challenges in post-genomic research is to develop methods able to extract information from the vast amount of data generated by high-throughput techniques (Church, 2005). Tools such as microarrays probing expression of all known genes are now a standard technique worldwide, whereas new approaches to measure expression based on ‘deep sequencing’ of total RNA are fast emerging (Sultan et al., 2008). This revolution involves also classical genetic studies: the use of high-throughput technologies in genetics is helping researchers to shed light on the complex genetic interactions underlying inherited phenotypes (Badano and Katsanis, 2002).
Such huge amount of information can be interpreted using methods derived from quantitative sciences such as physics, engineering and computer science. This new approach to research in Biology has come to be known as ‘Systems Biology’. Systems biology can proceed in two directions: a ‘bottom-up’ approach, in which starting from detailed knowledge of a biological process of interest, mathematical language is used to quantitatively describe the biological knowledge and the experimental data into a model of the biological process under study. The biological process is thus represented as a network describing interactions between genes, proteins, metabolites and other molecules. This network can then be used to probe the behavior of the biological process using computer simulations and mathematical analysis, to generate novel hypotheses to be then tested in vivo. The Systems biology ‘top-down’ approach, on the contrary, aims at learning the network of gene–gene interactions for a biological process for which very limited knowledge is available. This approach is called ‘reverse engineering’ and, typically, makes use of high-throughput gene expression profiling following a variety of perturbations to the cell to learn gene–gene interactions. Systems biology has contributed to the birth of another field, that of ‘Synthetic Biology’. This new discipline aims at constructing novel biological ‘circuits’ (synthetic networks) in the cell, using mathematical models to simulate their behavior and aid in their design, before their construction. The synthetic network is designed in such a way to perform a specific task of interest (for example, periodic expression of a gene of interest) (Elowitz and Leibler, 2000; Gardner et al., 2000) or to change the behavior of a natural occurring network in a desired way (for example, modify bacterial metabolism to produce a specific compound of interest) (Ro et al., 2006). Here we will review Systems and Synthetic biology approaches and examples of their application to genetics. We will also show how Synthetic biology is linked to this former science in the methods as well as in the aims (Figure 1). We will look at the relevance of this field and its approaches in biotechnology and in medicine. We will present selected practical applications, some of which only partially of Synthetic biology according to the mere definition, but still, in our opinion important to understand what will be possible to achieve in the near future, thanks to this new approach.

In this image we illustrate the general framework of Systems biology in which a model organism is perturbed and the data collected are used to understand novel biological functions. The results of this study are then fed back into the model to verify its predictive ability. By Systems biology studies, biological networks are better understood and divided into their single components that can then be used by Synthetic biology for the assemblage of novel circuits able to perform original functions. Circuits are modeled mathematically and models are used to improve the performance of the circuit.
Systems biology and reverse engineering
‘Reverse engineering’ is an ensemble of computational Systems biology approaches aimed at using gene expression data, or other available experimental data sets, to infer regulatory interactions among genes. There are two broad classes of reverse-engineering approaches according to Gardner and Faith (2005): those aimed at identifying ‘physical’ interactions between transcription factors (TF) and their target sequences from expression data (‘physical networks’), and those aimed at understanding the expression of related genes, even if the interaction does not involve any physical binding to DNA, or, a protein–protein complex (‘influence networks’). For more in-depth details we refer the readers to Gardner and Faith (2005) and Bansal and di Bernardo (2007). One of the first examples of reverse engineering from gene expression data was presented in Gardner et al. (2003). The authors studied a subset of nine genes in the SOS pathway of Escherichia coli, which is activated in response to DNA damage. They performed a set of nine transcriptional perturbations, where each perturbation consisted in the exogenous overexpression of only one of the nine genes of the network. The network inference by multiple regression (NIR) method (Gardner et al., 2003) was then applied to the expression data set obtained by measuring the response of the network genes to each of the perturbations. The NIR method is derived from a branch of engineering called system identification in which a model of the connections and functional relations between elements in a network is inferred from measurements of system dynamics (for example, the response of genes and proteins to external perturbations). NIR is based on multiple linear regressions to determine the model from RNA expression changes resulting from a set of steady-state transcriptional perturbations. The authors show that NIR correctly determined the circuit's wiring of the subset of nine genes in the SOS pathway. Recently, such reverse-engineering methods have been shown to be useful also in predicting mammalian gene networks. In what follows, we describe two examples of reverse-engineering application to infer mammalian transcriptional regulatory networks and recover direct targets of a TF, respectively.
Basso et al. (2005) inferred a global regulatory gene network in human B cells, using a novel method based on coexpression of genes across multiple data sets called ‘ARACNE’. The key idea being that two genes will change together in a collection of different gene expression profiles (microarrays) from the same cell type only if they are interacting. ARACNE works through identification of statistically significant gene–gene co-regulation by mutual information, an information-theoretic measure of correlation between two genes.
ARACNE was applied to genome-wide expression profiles from a panel of 336 B-cell phenotypes representative of a wide selection of normal, transformed and experimentally manipulated human B cells. A key result of this study was the ability to infer genetic interactions on a genome-wide scale from gene expression profiles of mammalian cells. In the inferred network, a relatively small number of highly connected genes (‘hubs’) interact with most other genes in the cell, either directly or hierarchically, through other highly connected subhubs. The proto-oncogene MYC emerged as one of the largest hubs in the network. ARACNE recapitulated known MYC target genes and identified new candidate targets, which were then validated by biochemical analysis. The success of ARACNE in the genome-wide identification of gene networks indicates that the method can be useful in the dissection of both normal and pathologic mammalian phenotypes.
In a more recent approach, Della Gatta et al. (2008) used an integrated computational and experimental approach to identify direct targets of Trp63 TF. This consists in measuring time-course (dynamic) gene expression profiles on perturbation of the TF, and in applying a novel reverse-engineering algorithm (Time Series Network Identification, TSNI) to rank genes according to their probability of being direct targets of Trp63. By applying TSNI to dynamic gene expression data following p63 activation, the authors were able to identify at least 53 direct and functional targets, including 39 novel targets, whereas 14 were already known.
TSNI is based on linear regression, where the observed changes in transcript concentration in time are assumed to be proportional to the concentration of the other transcripts in the cell, and to the inducible activation of the TF. One of the key messages of this work is that to capture the dynamics of gene expression in mammalian cells it is fundamental to consider short sampling times, in the order of minutes, whereas longer sampling times (hours) will miss relevant information (Della Gatta et al., 2008). TSNI takes into account the complexity of in vivo regulation of a direct target gene, such as feedback (or feed-forward) regulation, known to affect the timing of expression changes.
Systems biology and genetics
The use of high-throughput technologies together with Systems biology techniques could represent a valuable resource to study complex behaviors and to understand key genetic and protein interaction networks responsible for genetic diseases. Systems biology, thanks to high-throughput technologies, is helping modern geneticists to perform, on a larger scale, the same type of studies performed on a small scale using classical linkage studies (Brunner and van Driel, 2004; de Koning and Haley, 2005). Using powerful computational methods, it is now possible to integrate information maps deriving from different biological fields, thus helping in identifying candidate genes and understanding their functions (Beyer et al., 2007). Many examples of these new approaches to answer classical genetics questions can be found in recent literature.
Lage et al. (2007) applied a network-based approach for candidate gene identification. They performed a large-scale analysis of human protein complexes comprising gene products implicated in many different types of human disease to create a network of interactions and phenomena. In this analysis, they assumed that mutations in different members of a protein complex (predicted from protein–protein interaction data) led to comparable phenotypes.
Using a phenomic ranking of protein complexes linked to human disease, developed a Bayesian predictor that in 298 of 669 linkage intervals correctly ranks the known disease-causing protein as the top candidate, and in 870 intervals with no identified disease-causing gene provides novel candidates implicated in disorders such as Retinitis pigmentosa, epithelial ovarian cancer, inflammatory bowel disease, amyotrophic lateral sclerosis, Alzheimer's disease, type II diabetes and coronary heart disease. One more insightful example of a study based on protein–protein interaction networks can be found in Lim et al. (2006). They proposed a new phenotype-based protein–protein interaction approach to identify candidate genes for common diseases. Their approach is based on the hypothesis that the most relevant protein–protein interactions are conserved throughout evolution. By combining a stringent yeast two-hybrid screening with literature and evolutionary conserved interaction data, the authors constructed an interaction network of 20 proteins involved in the inherited ataxias disease.
Recently, two papers on the application of networks to the study of metabolic diseases were published. In the first work, Emilsson et al. (2008) took into consideration 23 270 expression data from various phenotypes, including clinical obesity. Through segregation analysis, genome-wide linkage and association studies they demonstrated an extensive genetic component underlying gene expression traits in blood and adipose tissue. In a similar way to the ARACNE method described above, they built a coexpression network to identify gene modules strongly associated with obesity, by considering all pair-wise correlation among the most differentially expressed genes detected in the tissue. Finally, after a comparison of human and mouse adipose tissue gene coexpression networks, they demonstrated an high conservation of key modules in human and in mouse, suggesting that inflammatory response and macrophage activation are very important pathways affected in this disease through evolution. In a companion paper, Chen et al. (2008), instead of identifying a single disease-susceptibility gene, as in classic forward genetics approaches, identified a subset of genes in the coexpression network that was affected by DNA variation associated with complex phenotypes such as obesity, diabetes and atherosclerosis. Integrating gene expression data generated from a segregating mouse population with genotypic data, and mapping them on the gene coexpression network, the authors identified a macrophage-enriched subnetwork that is associated with these metabolic syndromes and validated in vivo three genes (Lpl, lactb and Ppm1l) previously unknown to be obesity genes. The advantage of this approach is that the predicted network could help in identifying hundreds of genes that are disease responsible and that could serve as points for therapeutic intervention.
Synthetic biology
The words Synthetic biology appeared in literature for the first time in 1980, used by Barbara Hobom (Hobom, 1980) to describe genetically engineered bacteria. The discipline was then synonymous with genetic engineering; it was in fact the controlled manipulation of a gene in an organism with the intent of making that organism better or more useful in some way (for example, resistant to a specific molecule). The first generation of genetically modified organisms gave way later to more complex second- and third-generation GMOs, in which the better understanding of the genome and transcriptome allowed to introduce several interacting genes (Synthetic circuits).
During the year 2000, the engineering community got involved and began considering these new genetic circuits as electronic circuits, prone to being modeled mathematically and used in simulations whereas their single elements (genes) could be changed and moved around at will to obtain a desired behavior. This was Synthetic biology, as we will consider it throughout this review.
In their seminal work Gardner et al. (2000) present the construction of a genetic toggle switch that comprises two TFs that inhibit each other's expression. This reciprocal inhibition allows the toggle to be in one of two possible states, high TF1 and low TF2, or vice versa. Switching between the two is enabled by the transient introduction of an external inducer of the active repressor. Once the network has switched, the new epigenetic state is maintained indefinitely or until the application of the other inducer. This study includes a system of equations that describe the toggle behavior and the conditions necessary for bistability. As a practical device, the toggle switch is envisioned as a cellular memory unit; once equipped with it and induced with a first molecule the cell will ‘remember’ what gene is to be expressed until induction with a second external molecule.
In 2004, Kramer et al. (Kramer et al., 2004), building on their previous work carried out on the construction of novel inducible systems, constructed the first switch in mammalian cells, which has the same design as the one previously described and implemented by Gardner et al. (2000) in E. coli. The switch uses as promoters the streptogramin PpirON and the macrolide PetrON-inducible promoters, which direct expression of two repressors: E-Krab and PIP-Krab. Two antibiotics erythromycin and streptogramin are used as inducers to allow transition from state 1 to state 2. A system of differential equations was used to describe the circuits and to aid determining parameters allowing bistability. This work shows how it is possible to engineer epigenetic expression circuits for the maintenance of a desired relative level of proteins in mammalian cells. The toggle switch, in a gene therapy scenario, could be used to regulate in vivo the administration of a given gene product by switching on, or off, the expression of a gene with a transient treatment. For example, a patient may be treated with an antibiotic that works as an inducer for the switch, and once the switching has occurred, the treatment with the antibiotic could be suspended, but the protein will be produced indefinitely or until the application of the other inducer to switch the system off. More complex behaviors, such as periodic oscillations in the expression of a gene independently of the cell cycle, are also possible, even though real oscillations still have not being achieved. In one of the first examples of an oscillator (Elowitz and Leibler, 2000), the authors designed a circuit of three repressor genes in E. coli that turn each other off periodically and autonomously. The last gene activates a green fluorescent protein readout. The aim was to build an artificial clock that would oscillate with periods slower that cell-cycle division. In this work a system of equations describes a mathematical model of the network and is used to aid in determining how to tune the experimental parameters to obtain the desired oscillations. This work proves the predictability of a Synthetic circuit designed using nonlinear dynamic analysis. Such an oscillator has not yet been replicated in mammalian cells, but work is fast proceeding towards this end. An ongoing multipartner European funded project (www.cobios.net) aims at developing a mammalian oscillator to produce the insulin protein at specific time intervals from a population of engineered cells carrying the oscillator circuit in their genome. Recently, a Synthetic system for pattern formation in response to a variation in gradient was presented in Basu et al. (2005). The system takes advantage of the membrane diffusion of acylated homoserine lactone, which then binds LuxR, which in turns activates the expression of λ repressors (CI and Lac). Pattern formation is crucial in developmental processes and the hope is that this Synthetic approach will not only help to control and use stem cell differentiation and patterning but will also provide insight into the endogenous specification/differentiation mechanisms. This work is now being turned into a possible application in mammalian cells (ES cells) (R Weiss, personal communication). One of the applications envisioned is to develop a system regulating differentiation of pancreatic β-cells in function of cell density to enable a kind of ‘Synthetic’ homeostasis able to compensate autoimmune attacks in type I diabetes.
Synthetic biology approaches are now becoming even more integrated to Systems biology approaches with the aim to ‘rewire’ gene regulatory networks in living cells to generate a desired phenotypic behavior. This is a very challenging task and its first applications are being developed in prokaryotic cells with the aim of producing pharmacological compounds, or other important tasks in biotechnology, such as energy production.
One of the problems encountered by Synthetic biologists in building novel genetic networks performing desired functions in microorganisms, or in mammalian cells, lies in the fact that a large number of other genes are acting, at the same time, in the cell, and they often prove to have unexpected interactions with the new gene circuit. Hence groups of scientists are focusing on the production of cells with a minimal genome in which only the genes essential for the existence of the organism are expressed. To obtain minimal genomes it is either possible to adopt an evolutionary approach or to use systematic gene knockout. Progress is being made, and in 2006 Church published a detailed method to assemble a Synthetic cell from scratch (Forster and Church, 2006). It includes 115 genes, which combined with various biochemical agents permit a cell's life, if under carefully controlled lab conditions.
In Pal et al. (2006) the authors presented an approach in which they used an in silico representation of the metabolic system of E. coli to examine the role of each gene involved. They designed an algorithm that removes a randomly chosen gene in the network; it then calculates the impact on the deletion on the production rate of biomass components (proxy for fitness) and finally removes the gene if the impact of deletion is negligible, or keeps it if the impact is significant. This process is repeated for all the genes in the network for 500 times to simulate evolution. The networks obtained shared 77% of their reactions compared to only 25% when genes were deleted randomly. Simulations were then performed on Buchnera aphidicola and Wigglesworthia glossinidia, probably result of reductive evolution, close relatives of E. coli, and for both the species the network model accuracy was increased compared to the one expected by chance.
Craig Venter and his team worked for years to develop a minimal genome containing less than 400 genes, which has everything it takes to allow life (Hutchison et al., 1999). In this work, Venter and his team investigated which genes would be essential for life by knocking out genes in the bacterium Mycoplasma genitalium, an organism that has a very small genome (about 400 genes) and ended up concluding that only between 250 and 350 genes were crucial under laboratory conditions. Their next step was to synthesize from scratch the minimal genome, and put it into a bacterial cell (Gibson et al., 2008).
In Ro et al. (2006) we can find a practical application of genetic engineering, in a scent of Synthetic biology, applied to a microorganism. The authors re-engineered a strain of yeast that produces artemisinic acid, a precursor of artemisinin, a compound normally extracted from Artemisia annua (sweet wormwood) and highly effective against malaria. In their work, the group increased the production of farnesyl pyrophosphate (FPP) a compound already produced in Saccharomyces cerevisiae, which acts as a precursor in the pathway for the biosynthesis of artemisinin in sweet wormwood. They then proceeded to modify the yeast genome adding amorphadiene synthase, a gene from A. annua that produces a catalyzer for the circularization of FPP molecule to amorphadiene and finally cloned a novel cytochrome P-450 that oxidizes amorphadiene to artemisinic acid. The final result was a yeast strain capable of producing artemisinic acid at a biomass comparable to that produced by A. annua; this study lacks the mathematical modeling that in this review we consider as a fundamental part of the definition of Synthetic biology, but it does show a prime example of modification of a natural organism pathway to obtain a novel and useful function. In this sense it can be considered to have a Synthetic biology scope, as it was able to ‘rewire’ a biological process in a desired way.
Synthetic biology in medical applications
Medical applications of Synthetic circuits have also been proposed, but the field is still in its infancy. Fux et al. (2001) in their work present the construction of a circuit to control growth in mammalian cells. This work focuses on the fact that to have success in cells and tissue engineering it will be necessary to manage temporal proliferation of cells. In tissue engineering a number of disciplines including biology, medicine and engineering are used in synergy to reinstate, maintain and at times enhance tissue and/or organ function. The foundation of this discipline is the ability to exploit living cells for diagnostic applications and to test drug metabolism; it is therefore fundamental to have populations of cells for which proliferation is rigorously controlled. To this end Fux et al. (2001) engineered different inducible circuits and tested them in Chinese hamster ovary (CHO) cells. The use of the two promoters PhCMV and Ppir, respectively, from the TetOFF and the PipOFF Systems allows for dual gene regulation with doxycycline and streptogramin. A range of combination of these two promoters with the human-cyclin-dependent kinase inhibitor p27Kip1, whose overexpression arrests CHO cells in G1 phase, and the growth-promoting SV40 small T antigen, was explored. A perfect control of growth and proliferation was only achieved by expression of p27Kip1 in sense and antisense, respectively, under the control of the tetracycline-inducible promoter and of the streptogramin-inducible promoter. This work is of interest because it shows an early age attempt at Synthetic biology with a clear aim. In a recent work, Anderson et al. (2006) proposed to design bacteria to be used as targeted delivery systems for a possible strategy of cancer therapy. inv is a gene from Yersinia pseudotuberculosis encoding invasin, a protein that permits adhesion and invasion of mammalian cells. In this study the cited gene is inserted in an E. coli strain that lacks endogenous TetR under the TET promoter to obtain constitutive expression and invasion of HeLa cells. inv is then put under the control of the araBAD operon to obtain arabinose-inducible invasion, and of fdhF to invade only in presence of hypoxic environment (often associated with tumor formation) and under LuxI promoter to elicit a quorum sensum response. This work is of major interest because it foresees a practical application of Synthetic biology; again it lacks a mathematical approach.
Discussion
Traditional genetics has focused on identifying single genes whose mutations are responsible for specific diseases. In the past 10 years, the paradigm has shifted from single genes to gene networks: genes, protein and all the other biomolecules regulate each other in a complex interaction network. One of the major challenges for geneticists and biologists, in the post-genomic era, is to find new methods to extrapolate correct and reliable information on gene interactions from huge amount of data, produced by high-throughput technologies. Modern biology can now be considered in the holistic sense; an approach to research that emphasizes the study of complex systems seen as networks of interconnected components, whose collective behavior yields phenotypes that cannot be understood by studying each component separately. This practice is alternative to reductionism, a purely analytic tradition, that purports to understand systems by dividing them into their smallest possible or discernible elements and understanding their elemental properties alone. Over the past 30 years, classical genetic studies of multifactorial human diseases successfully identified some of the genes and their allelic variants that can be considered irrefutable or true positives. However, there are probably hundreds of susceptibility loci that increase the risk for each common disease to be discovered. The emerging network-based approach to the identification of sets of genes responsible for increased risk in a specific disease is likely to speed up genetic research.
Systems biology approaches are particularly promising to discover protein key modulators in inherited diseases and to study complex genetic interaction that underlie most inherited phenotypes.
Synthetic biology can be considered as the other side of the coin of Systems biology, as it aims at making use of the naturally occurring biological components and at ‘re- engineering’ them to perform novel functions. Understanding how emerging properties can be generated from biological networks is necessary to be able to build ‘de novo’ synthetic networks to give rise to the desired phenotype (that is, oscillatory behavior or switch). This is the reason why Systems and Synthetic biology need each other to be successfully applied. Even though Synthetic biology is still very much at the stage of finding its definitions and testing prototypes, many research groups are turning to it. It is a very promising field both because of its mere applications and the tools it can provide, and progress is proceeding fast. The main challenges lie in the length of the process of assemblage of new DNA sequences (Endy, 2005), and in the fact that the main applications still lack mathematical models. In this review we highlighted the studies lacking a formal model, and it goes to show that there is still a certain miscommunication between modeling and practice. Synergy between biology and engineering is not easily achieved. Nevertheless the new scientist, that is the Synthetic biologist, could achieve goals not permitted to classical biologists, because he/she uses new interdisciplinary tools. So far, Synthetic biology dealt with the synthesis of novel polynucleotides and of new proteins, the redesigning of pathways and the creation of standard bioparts, as well as the study of the minimal genome required to an organism for life. These studies provided essential insight into known biological processes and will in the future most likely develop into promising applications. Thanks to ex vivo cell therapy, improved viral vector technology, and to a standardized characterization of the molecular building blocks (biobricks), in a few years from now we could have controllable gene therapy and be able to build large networks of genes with desired functions. The applications and the potential are immense and they are leading scientist onto unexplored grounds.
References
Anderson JC, Clarke EJ, Arkin AP, Voigt CA (2006). Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol 355: 619–627.
Badano JL, Katsanis N (2002). Beyond Mendel: an evolving view of human genetic disease transmission. Nat Rev Genet 3: 779–789.
Bansal M, di Bernardo D (2007). Inference of gene networks from temporal gene expression profiles. IET Syst Biol 1: 306–312.
Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A (2005). Reverse engineering of regulatory networks in human B cells. Nat Genet 37: 382–390.
Basu S, Gerchman Y, Collins CH, Arnold FH, Weiss R (2005). A synthetic multicellular system for programmed pattern formation. Nature 434: 1130–1134.
Beyer A, Bandyopadhyay S, Ideker T (2007). Integrating physical and genetic maps: from genomes to interaction networks. Nat Rev Genet 8: 699–710.
Brunner HG, van Driel MA (2004). From syndrome families to functional genomics. Nat Rev Genet 5: 545–551.
Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ et al. (2008). Variations in DNA elucidate molecular networks that cause disease. Nature 452: 429–435.
Church GM (2005). From systems biology to synthetic biology. Mol Syst Biol 1: 1–2, 2005.0032. doi:10.1038/msb4100007.
de Koning DJ, Haley CS (2005). Genetical genomics in humans and model organisms. Trends Genet 21: 377–381.
Della Gatta G, Bansal M, Ambesi-Impiombato A, Antonini D, Missero C, di Bernardo D (2008). Direct targets of theTRP63 transcription factor revealed by a combination of gene expression profiling and reverse engineering. Genome Res 18: 939–948.
Elowitz MB, Leibler S (2000). A synthetic oscillatory network of transcriptional regulators. Nature 403: 335–338.
Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J et al. (2008). Genetics of gene expression and its effect on disease. Nature 452: 423–428.
Endy D (2005). Foundations for engineering biology. Nature 438: 449–453.
Forster AC, Church GM (2006). Towards synthesis of a minimal cell. Mol Syst Biol 2: 45.
Fux C, Moser S, Schlatter S, Rimann M, Bailey JE, Fussenegger M (2001). Streptogramin- and tetracycline-responsive dual regulated expression of p27(Kip1) sense and antisense enables positive and negative growth control of Chinese hamster ovary cells. Nucleic Acids Res 29: E19.
Gardner TS, Faith JJ (2005). Reverse-engineering transcription control networks. Phys Life Rev 2: 65–88.
Gardner TS, Cantor CR, Collins JJ (2000). Construction of a genetic toggle switch in Escherichia coli. Nature 403: 339–342.
Gardner TS, di Bernardo D, Lorenz D, Collins JJ (2003). Inferring genetic networks and identifying compound mode of action via expression profiling. Science 301: 102–105.
Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J et al. (2008). Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319: 1215–1220.
Hobom B (1980). Surgery of genes. At the doorstep of synthetic biology. Medizin Klinik 75: 14–21.
Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM et al. (1999). Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286: 2165–2169.
Kramer BP, Viretta AU, Daoud-El-Baba M, Aubel D, Weber W, Fussenegger M (2004). An engineered epigenetic transgene switch in mammalian cells. Nat Biotechnol 22: 867–870.
Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O et al. (2007). A human phenome–interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25: 309–316.
Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF et al. (2006). A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125: 801–814.
Pal C, Papp B, Lercher MJ, Csermely P, Oliver SG, Hurst LD (2006). Chance and necessity in the evolution of minimal metabolic networks. Nature 440: 667–670.
Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM et al. (2006). Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440: 940–943.
Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M et al. (2008). A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cuccato, G., Gatta, G. & di Bernardo, D. Systems and Synthetic biology: tackling genetic networks and complex diseases. Heredity 102, 527–532 (2009). https://doi.org/10.1038/hdy.2009.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.18
Keywords
This article is cited by
-
Synthesizing genetic sequential logic circuit with clock pulse generator
BMC Systems Biology (2014)
-
A conceptual review on systems biology in health and diseases: from biological networks to modern therapeutics
Systems and Synthetic Biology (2014)
