
Abstract
Dissection of complex traits that influence fitness is not only a central topic in evolutionary research but can also assist breeding practices for economically important plant species, such as loblolly pine (Pinus taeda L). In this study, 46 single nucleotide polymorphisms (SNPs) from 41 disease and abiotic stress-inducible genes were tested for their genetic association with carbon isotope discrimination (CID), a time-integrated trait measure of stomatal conductance. A family-based approach to detect genotype/phenotype genetic association was developed for the first time in plants by applying the quantitative transmission disequilibrium test on an association population of 961 clones from 61 families (adopted from previous breeding programs) evaluated for phenotypic expression of CID at two sites. Two particularly promising candidates for their genetic effects on CID are: dhn-1, involved in stabilization of cell structures, and lp5-like, a glycine rich protein putatively related to cell wall reinforcement proteins, both of which were shown in previous studies to be water-deficit inducible. Moreover, association in lp5-like involves a nonsynonymous mutation in linkage disequilibrium with two other nonsynonymous polymorphisms that could, by acting together, enhance overall phenotypic effects. This study highlights the complexity of dissecting CID traits and provides insights for designing second-generation association studies based on candidate gene approaches in forest trees.
Similar content being viewed by others
Introduction
Candidate gene association studies aim at linking phenotypic variation with allelic variation in candidate genes and benefit from several generations of recombination in natural populations to identify causative polymorphisms (reviewed in Cardon and Bell, 2001; Gupta et al., 2005; Hirschhorn and Daly, 2005; Laird and Lange, 2006; see Neale and Savolainen, 2004 for conifers). In plants, association studies have been relatively successful, but only a limited number of genes and traits have been tested for association to date. In forest trees, only two studies are available: Thumma et al. (2005) identified allelic variation in cinnamoyl CoA reductase affecting microfibril angle, a wood quality trait, in Eucalyptus and González-Martínez et al. (2007) found four genes (cad, sams-2, lp3-1 and α-tubulin) associated with different wood property traits in Pinus taeda. Most genetic association studies so far have targeted major commercial traits (wood property traits in commercial forest trees, Thumma et al., 2005; González-Martínez et al., 2007; kernel composition, starch properties and forage digestibility in maize, Guillet-Claude et al., 2004; Wilson et al., 2004) or focused on well-known pathogen resistance or flowering time genes (Thornsberry et al., 2001; Aranzana et al., 2005; see also Zhao et al., 2007). Addressing traits of a higher complexity, such as those drought related, may pose additional difficulties.
All previous examples of association studies in plants have, without exception, focused on natural populations. In contrast, association studies on other organisms such as humans and cattle have normally used family-based populations (Hirschhorn and Daly, 2005; Laird and Lange, 2006). Family-based designs in association studies might incorporate advantages of both linkage-based and linkage disequilibrium-based quantitative trait dissection approaches (for example, the transmission disequilibrium test (TDT) and its multiple extensions; Spielman et al., 1993; see review in Laird and Lange, 2006). Families might be generated through controlled crosses among a diverse selection of unrelated individuals according to a breeding scheme that aims at shuffling of alleles in multiple samples either across backgrounds or against a reference background, thus enhancing the level of linkage disequilibrium (LD) observed in the parents (Yu et al., 2006). The subsequent generations of progeny of the crosses can then be used as association populations (reviewed in Ersoz et al., 2007).
Admixture and stratification are known biases in genetic association studies and a major cause of false-positives in classic studies based on natural populations (see, for instance, Hirschhorn and Daly, 2005; Figure 3). However, attempted corrections of false positives produced by population structure can result in removing true-positives, that is, causative polymorphisms that are removed because they are strongly correlated with population structure. For example, Zhao et al. (2007) noted that correcting false positives produced by population structure in Arabidopsis removed many of the best candidates for extreme late-flowering phenotypes of northern accessions (northern Sweden and Finland), which were genetically different from the others. Indeed, any polymorphism shared by these accessions and giving positive association with a flowering-time phenotype would be considered a false positive due to the misleading effect of population structure. In contrast to methods for association based on natural populations, family-based methods are robust against population admixture and stratification. Despite their obvious advantages, standard TDTs, such as quantitative TDT (QTDT) (Abecasis et al., 2000a, 2000b), require individuals to have heterozygous ancestors in the pedigree to be informative and might have lower power than classic natural population-based association studies.
The response of plants to dehydration is complex and involves numerous biochemical, physiological and morphological alterations to reduce water loss and protect cells from desiccation (reviewed in Ingram and Bartels, 1996; Shinozaki and Yamaguchi-Shinozaki, 2007). Abiotic stress-inducible genes include: (i) signaling cascades (Dubos and Plomion, 2003, ii) transcription factors, either ABA-independent (for example, DREB2, Riera et al., 2005; Agarwal et al., 2006) or ABA-dependent (for example, MYB, Cominelli et al., 2005; Riera et al., 2005); (iii) protection factors of macromolecules (for example, dehydrins and chaperones, Close, 1997; Ismail et al., 1999; Rorat, 2006); (iv) detoxification enzymes (for example, antioxidants, Karpinska et al., 2001; Reddy et al., 2004) and (v) water channels and transporters (for example, aquaporins, Luu and Maurel, 2005; Kiani et al., 2007). In natural populations of forest trees, some of these genes are likely adaptive (for example, ccoaomt-1 and erd3 in Pinus taeda, González-Martínez et al., 2006; pr-agp4, erd3, dhn-1, dhn2 and lp3-1 in Pinus pinaster, Eveno et al., 2008). Remarkably, gene expression of drought-induced genes can vary even in populations located in close geographic proximity (Sathyan et al., 2005), which might reflect different adaptations to drought tolerance, as it has already been shown for physiological traits in experimental conditions (for example, Nguyen-Queyrens and Bouchet-Lannat, 2003).
The complexity of drought-response in plants also poses difficulties for measuring drought-related traits. Several parameters related to the hydraulic properties of trees have been suggested, such as leaf-to-wood area ratios, leaf hydraulic conductivity, vulnerability to xylem embolism, carbon isotope discrimination (CID) of needles/leaves or wood (that is, the ratio between stomatal conductance and photosynthetic capacity; Farquhar et al., 1989) and different water potentials (reviewed in Martínez-Vilalta et al., 2004 for Pinaceae). Among all these methods, CID (Δ) has been favored in recent years because of its amenability to high-throughput phenotyping and its putative correlation with other hydraulic parameters, at least in Pinaceae (Martínez-Vilalta et al., 2004 and references therein). Furthermore, genetic studies from model organisms such as Arabidopsis indicated that CID might have higher heritability in C3 plants than other measures of WUE (McKay et al., 2003). Nevertheless, CID is a complex trait that can also vary substantially with environmental variables, such as altitude (see, for example, Warren et al., 2001) or CO2 concentration (Tjoelker et al., 1998). Warren et al. (2001) also found that CID was notably affected by both fertilization levels and stand density in Pinus radiata and P. pinaster. Therefore, care must be taken when interpreting CID results in terms of drought tolerance (see also Baltunis et al., 2008).
Testing and improving tree breeding stocks for enhanced drought tolerance and increased water use efficiency (WUE) have become major objectives in commercial production, especially considering recent (and future) global climate changes. Dissecting the molecular basis of drought tolerance in plants also has relevant implications for conservation genetics and evolutionary research. In this paper, 46 single nucleotide polymorphisms (SNPs) from 41 disease and abiotic stress-inducible genes, previously screened for their patterns of nucleotide diversity and LD (Brown et al., 2004; Ersoz, 2006; González-Martínez et al., 2006), were tested for association with CID, a trait related to WUE and potentially to drought, in forest trees.
Materials and methods
Plant material and trait measurements
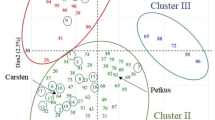
Sixty-one families were generated using a partial diallel mating design as part of the Forest Biology Research Cooperative (FBRC) Tree Improvement Program (University of Florida, Gainesville, FL, USA), using 31 diverse natural selections (Figure 1) from three provenances (Atlantic Coastal Plain, Florida and Gulf Coast) that were previously analyzed for their nucleotide diversity and LD levels (Brown et al., 2004; Ersoz, 2006; González-Martínez et al., 2006). Family size varied from 15 to 18 clones, with an average of 16. Propagules were allocated to replications according to an incomplete block design with 12–16 trees per incomplete block, in each of two sites (Cuthbert, Georgia and Palatka, Florida). Full common garden design description and details on isotope ratio measurements are given in Baltunis et al. (2007, 2008). Measurements were repeatable and accurate with a standard error of 0.14‰. CID values (Δ) were calculated from δ13C values using Equation (1) (Farquhar et al., 1989):

where δp is the isotope composition of the plant material and δa is that of the air (assumed to be −8‰).

Origin of parental first-generation selections and location of study sites (stars). Size of dots is proportional to the number of trees sampled at that location. Minimum temperature isotherms indicating zones recommended for seed transfer are also shown (see Schmidtling, 2001). The shadowed pattern represents the native range of P. taeda in southeastern United States.
Mean discrimination at the Georgia and Florida sites was 21.2 and 19.6‰, respectively. Provenance and family effects were significant, CID being slightly more heritable at Cuthbert than Palatka, with narrow-sense heritability (h2) estimates of 0.20 and 0.14, respectively, and an across-site estimate of 0.09 (Baltunis et al., 2008). Correlation across sites was only moderate, probably due to environmental differences between sites and genotype per environment (G × E) interactions. G × E interactions and the contrasting architecture of the nonadditive genetic variance at Cuthbert and Palatka sites (due to epistasis and dominance, respectively; see Baltunis et al., 2008) argue in favor of treating CID in the two sites as different traits. A linear model was used to generate best linear unbiased predictions for each single site as follows:

where μ is the overall mean; A, B and C are regressors with values equal to 0, 0.5 or 1 depending on whether the parents for a given family belongs to the same provenance (for example, A=1, B=0, C=0) or not (for example, A=0.5, B=0.5, C=0), thus representing the provenance effect; ri is the i-th repetition; gcaj and gcak are the general combining ability of the j-th female and the k-th male, respectively; scajk is the specific combining ability of the j-th female with the k-th male; c(fam)jkl is the l-th clone within the jk-th family and eijkl is the error term. The ri, gcaj, gcak, scajk and c(fam)jkl are treated as random variables. Clone best linear unbiased predictions for genotype/phenotype genetic association were generated by summing the provenance estimates and the gcaj, gcak, scajk and clone within family, c(fam)jkl, predictions. Analyses were performed using SAS 9.0 and ASReml statistical packages.
DNA isolation and SNP genotyping
DNA isolation was performed by grinding needle tissue in liquid nitrogen followed by whole DNA extraction using the QIAGEN DNeasy Maxi plant DNA extraction kit (Valencia, CA, USA). A total of ∼5–10 ng of DNA was used for down stream PCR applications. Genotyping was conducted on a Victor2-Wallac SNP genotyping platform with the AcycloPrime Universal Fluorescence Polarization Terminator Dye Incorporation kit (FP-TDI, Perkin-Elmer, Torrance, CA, USA; see Kwok, 2002, for a description of the method). PCR reactions were conducted following the manufacturer's AcycloPrime FP-TDI assay protocols and adjusting dNTPs (200–800 μM) and primers (200–800 nM) concentration and number of PCR cycles (15–35). Sequences for the genotyping primers, their annealing temperatures, direction of single-nucleotide extension reaction, minor allele frequency (MAF) and the alleles at the SNP loci are listed in Supplementary Table S1. A total of 46 SNPs were genotyped in 961 clones from 61 families, resulting in ∼44 206 data points, with ∼10% missing data.
Genetic association methods
Several family-based methods have been developed in the last decade, starting with the original formulation of the TDT by Spielman et al. (1993). Here, the orthogonal model of Abecasis et al. (2000a, 2000b) was used for data analysis (see also Fulker et al., 1999). This extension of the TDT for quantitative traits, the QTDT, is based on the difference between average phenotypic values of individuals with different alleles transmitted from a heterozygous parent, computed using standard variance-component methods and the identity by descent among relatives. The QTDT is robust to population stratification and admixture. First, for a bi-allelic marker M with arbitrarily named alleles A and B, we define the genotype score gij for the j-th offspring in the i-th family as the number of ‘A’ alleles at locus M minus one. Second, the genotype score (gij) is decomposed into two orthogonal components: the between-family component bi and the within-family component wij=gij−bi. In this formulation, bi represents the average within-family genotype that in our study case is obtained from sib information as follows:

Then, the means model under this specification is:

where μ is the overall mean; bi and wij are the orthogonal between- and within-family components of gij; and βb and βw are regressors. Within-family tests of association are developed by comparing quantitative models, including only the between-family component (null model) or both within- and between-family components (full model), using likelihood ratio tests that assume a normal distribution for the traits. In the absence of population structure, total association confers more power and makes detection of correlation between a marker and an underlying trait easier than within-family association. Therefore, we also tested for total association, using both within-family and between-family components, in the cases where no evidence of population stratification was observed (βb=βw). Finally, a Monte-Carlo permutation framework was used to compute unbiased P-values. This permutation scheme corrects for small sample sizes, ascertainment bias and, most importantly, for deviations from normality of phenotypic variables. Similar results to the orthogonal model were obtained using the Monks et al. (1998) model and are thus not presented here. Analyses were done using QTDT software (available at www.sph.umich.edu/csg/abecasis/QTDT/; January 2008).
When significant genetic association was observed, the approximate phenotypic variation explained by the marker was calculated as:

where Vp is the total phenotypic variance, p is the marker allele frequency and a is the additive effect, which is estimated by the βw regression coefficient (Fulker et al., 1999; Abecasis et al., 2000a).
Data perturbation simulations
Following Yu et al. (2006) and Zhao et al. (2007), a simulation scheme based on the perturbation of existing phenotypes was implemented. The method consisted in the addition of a constant additive effect to the minor allele of a randomly chosen causal SNP, while keeping the real data structure. Allelic fixed effects ranging from 0.1 to 0.5 times the standard deviation of the phenotype (accounting for SNP effects on phenotype of ∼0.5–10%) were considered. Simulations were used to compare the power of QTDT and the family-based design used here with an unstructured population of the same size (N=961), generated from the sibs allele frequencies and tested for association using standard general linear models. Power estimates were computed separately for different MAF classes (MAF<0.1, 0.1⩽MAF<0.2 and MAF⩾0.2).
Results
Quantitative transmission disequilibrium test was used to obtain evidence for association between SNP alleles in 41 candidate gene loci to CID phenotypes at two field trials in which the association population was grown. CID was heritable at both sites (H2=0.33 at Cuthbert, GA and 0.25 at Palatka, FL; Baltunis et al., 2008). Analyses for within-family genetic association were based on an average of 415 and 495 probands (that is, sibs with heterozygous parents) in Cuthbert and Palatka, respectively. In general, SNPs giving genetic association at the within-family level were also significant or nearly significant for total association (only SNPs that did not show population structure were tested for total association; see Table 1).
Significant genetic association was found between CID phenotypes measured at Cuthbert and the silent SNP Q1 in 44 segregating families within the dehydrin 1 (dhn-1) locus. At the Palatka site, significant genetic association was found for C13 in 26 segregating families; C13 is a nonsynonymous polymorphism in a putative cell wall protein similar to lp5 in Pinus taeda. Another two (silent) SNPs were significantly associated with CID phenotypes: C22 (18 segregating families) from a wrky-like transcription factor and S9 (32 segregating families) from Cu/Zn superoxide dismutase (sod-chl) gene at Cuthbert and Palatka, respectively, but supporting evidence for genotype/phenotype associations was weaker (Table 1). No significant associations between candidate SNPs and CID phenotypes were detected simultaneously at both sites. This is consistent with the moderate genetic correlation for the CID trait across sites, which indicates rank changes in the expression of the CID phenotype by clones at the two sites. Thus, we did not necessarily expect identical associations to be detected for both sites in this analysis.
None of these associations were significant after correcting for multiple testing (Bonferroni threshold for P<0.05 was 0.001) and the allelic effect on the phenotype was lower than 1% in all cases, except one (C22, which explained 3.38% of the phenotypic variance), as roughly estimated by (S). Weak associations (0.5<P⩽0.10) were detected for chromatin assembly transcription factor 1 (caf-1), caffeoyl-CoA-O-methyltransferase 1 (ccoaomt-1), ethylene insensitive 2 (eins-2) and another wrky class transcription factor (Supplementary Table S2). Finally, (more powerful) tests of total association but not within-family tests pointed out tentative genetic association for a terpene synthase-like (tps-like) gene in Cuthbert and a myb class transcription factor in Palatka (see Supplementary Table S2).
Simple simulations showed a reasonably high power to detect association of QTDT with the present family-based experimental design given frequent SNPs (MAF⩾0.2) and allelic effects of ∼0.1 times the phenotypic standard deviation (equivalent to ∼1% of explained phenotypic variance per SNP) (Figure 2). However, to reach significance levels after correction for multiple testing (Bonferroni threshold for P<0.05 was 0.001), higher allelic effects, of the order of ∼2.5% of explained phenotypic variance per SNP, were needed. Power to detect association decayed rapidly for low frequency (0.1⩽MAF<0.2) and rare alleles (MAF<0.1) (see Figure 2). In comparison with an equivalent design based on unstructured populations (and analyzed using standard general linear models), QTDT and the family-based design used in this study achieved less power in all scenarios. Differences between methods were especially notable when low frequency and rare alleles were considered.

Power (as estimated by P-values achieved) of the family-based design used here (dark lines) in comparison with an unstructured population of the same size (N=961), generated from sibs allele frequencies (light lines) and tested for association using standard general linear models (GLMs). Estimates are presented separately for different minor allele frequency (MAF) classes (MAF<0.1, 0.1⩽MAF<0.2 and MAF⩾0.2).
Discussion
Response to and recovery from drought is a complex process that involves multiple biochemical, physiological and morphological adaptations in plants. Pines are particularly vulnerable to xylem embolism (reviewed in Martínez-Vilalta et al., 2004) and have developed multiple mechanisms both to avoid and to tolerate drought, which are commonly recognized (see, for instance, Table 1 in Newton et al., 1991). In this study, positive genetic association between a number of drought-inducible candidate genes and CID in needles was found. The CID, applied to different plant tissues (generally needles or leaves but also wood), is a time-integrated estimator of photosynthetic WUE, that is, the ratio between net CO2 assimilation and stomatal conductance, and has been extensively used to compare response to drought in plants, including forest trees (reviewed in Warren et al., 2001; Dawson et al., 2002; Adams and Kolb, 2004; Monclus et al., 2006).
High variation in CID has been found across species (for example, Oliveras et al., 2003 for conifers; Cernusak et al., 2007 for tropical trees), as well as moderate narrow-sense (h2) heritability within-species (0.17 in maritime pine, Brendel et al., 2002; 0.09 across-sites in loblolly pine, Baltunis et al., 2008). As trees with higher WUE may sustain growth under water-limitation conditions and differences in WUE represent also different growing strategies, CID is an attractive trait for breeding, in particular in dry areas or in those regions in which the higher impact of the current process of global climate change will be felt.
Genotype/phenotype associations involve four genes belonging to different functional classes related to drought: general protection factors (dhn-1), antioxidants (sod-chl), transcription factors (wrky-like) and putative cell-wall proteins (lp5-like). Dehydrins are accumulated in vegetative tissues in response to cell dehydration and multiple protection-related functions have been described in numerous organisms for this widespread gene family (for example, stabilization of vesicles or other endomembrane structures, metal-binding activity, protection from oxidative damage, cryoprotective activity, and so on; see reviews in Close, 1997; Allagulova et al., 2003; Rorat, 2006). Dehydrins can be classified in structural types, probably related to distinct functions (Rorat, 2006), attending to the number and order of different domains (named the Y, S and K segments; see details in Close, 1997). Dhn-1 showed 85% similarity to PgDhn-1 from Picea glauca, a SKn-type dehydrin that is overexpressed under wounding, cold and drought stress (Richard et al., 2000). SKn dehydrins have been suggested to be involved in cold acclimation (Rorat, 2006 and references therein), metal detoxification (Zhang et al., 2006) and other abiotic-stress responses (Zhang et al., 2007).
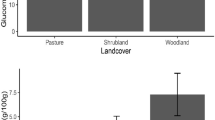
Another candidate gene giving positive association with CID was a transcription factor, related to the WRKY family (that is, transcription factors that contain a DNA-binding region comprising the conserved sequence motif WRKY adjacent to a zinc-finger motif). Although little is known about this transcription factor family, their members seem to be upregulated by wounding, pathogen infection and diverse abiotic stresses, such as cold or drought (see reviews in Eulgem et al., 2000; Ülker and Somssich, 2004). Interestingly, the WRKY transcription factor family is involved in defense-induced mitogen-activated protein kinase signaling cascades (Asai et al., 2002) and in leaf senescence (Robatzek and Somssich, 2002) in Arabidopsis. The only candidate gene showing genetic association in both common garden experiments (albeit only marginally in the Cuthbert trial) was sod-chl, a Cu/Zn superoxide dismutase gene. At the Palatka site, CID of CT genotypes at sod-chl S9 was higher than TT genotypes in 70% of the families, with a mean within-family difference of 0.0551, while at the Cuthbert site the average difference between genotypes was around half this value (0.0298 with higher CID for CT in 63% of the families). Antioxidant activity increases after drought stress-induced generation of active oxygen species, which attack sensitive macromolecules resulting, ultimately, in cell death. Superoxide dismutases, in particular, are key players in the antioxidant defense system of most organisms, including humans (see, for instance, Wang et al., 2003; Reddy et al., 2004). The only significant genetic association involving a nonsynonymous polymorphism (C13) was found in a putative cell wall protein, similar to lp5 in Pinus taeda (Figure 3a; see Chang et al., 1996 for a description of lp5 gene), which also has a 48.92% similarity with a glycine-rich protein in Arabidopsis (accession number O65450). Glycine-rich proteins are often associated with stress response in plants (reviewed in Mousavi and Hotta, 2005), as it may also be the case for lp5 (see expressional profile in Figure 3c). Nonsynonymous SNP C13 (highlighted in Figure 3b) is in LD with two other nonsynonymous mutations (not tested for association in this study) located toward the 5′ end of the coding region. Multiple nonsynonymous mutations acting together (through LD) in the same direction may have higher phenotypic effects than single mutations.

Putative cell-wall protein lp5-like: (a) protein alignment with P. taeda's lp5 (AAB66348; reference sequence on top) highlighting nonsynonymous polymorphisms (including C13, S → R); (b) linkage disequilibrium plot (D′ and r2 are shown in the upper and lower parts of the plot, respectively, as obtained from Tassel software) for nonsynonymous single nucleotide polymorphisms, showing total and partial linkage of C13 with other nonsynonymous mutations close to the beginning of the coding sequence and (c) expressional profile obtained from in silico analysis using the Magic Gene Discovery tool (Laboratory for Genomics and Bioinformatics, University of Georgia; www.fungen.org); the pie graph represents the percentage of lp5-like ESTs (expressed sequence tags) found in root cDNA libraries from pines submitted to different stress treatments (nutrient deficiency, dark and abiotic stress) and in control libraries built with nonstressed pines (see further details in Lorenz et al., 2006).
The associations between CID and drought-inducible genes could be interpreted as reflecting adaptation to water-limiting conditions. However, water was probably not limiting for growth at either of the two loblolly pine testing sites, as indicated by a high water table at the time during which sampled leaves developed at the Palatka site and the positive correlations between growth and CID across both sites (Baltunis et al., 2008). Thus, associations might be generated through differences in photosynthetic capacity, for example, through protection of the photosynthetic apparatus by dehydrins, during transient periods of water deficit that might occur in foliage during peak temperatures. Alternatively, the associations may be driven by the action of genes in LD with the detected polymorphism. Further analysis with a much larger set of candidate genes will answer this question. In addition, validation of significant associations between CID and candidate genes in other association populations tested in stressed and nonstressed environments will be required to establish a more meaningful biological relationship between CID and drought-responsive genes.
None of the significant associations found in this study explained a substantial amount of the phenotypic variance present in the CID trait (<1% in all cases except C22) and only slightly higher values have been reported in other tree association studies, even for well-known traits such as those related to wood properties (Thumma et al., 2005; González-Martínez et al., 2007). This fact may indicate genetic control by many loci with relatively small individual effects and probably complex gene interactions. In addition, generally low phenotypic effects of single genes might explain the lack of significant association between CID phenotypes and some of the most promising drought-tolerance candidate genes tested in this study (such as dhn-2 or erd3).
This study highlights the complexity of WUE in trees and provides insights for designing second-generation association studies for drought tolerance. First, CID, a commonly measured trait related to WUE, showed a remarkable environmental influence. This fact argues in favor of strict environmental controls and testing in a wide range of environments and water-deficit conditions. Second, genetic associations at Cuthbert and Palatka sites involved different functional candidate gene types, highlighting the multiplicity of drought response mechanisms in plants and the complexity of compose traits such as CID. These complexity, as compared with relatively simple and well-known metabolic pathways (for example, the lignification pathway, see Peter and Neale, 2004) and traits, might complicate validation of genotype/phenotype associations for WUE in future studies. It also argues for a wide sampling of the genome to cover a variety of processes, such as: water channeling, signaling cascades, radical scavenging, macromolecules and membranes stabilization, and so on. Finally, given the low variance explained by most associations (<5%, see also González-Martínez et al., 2006) and the relatively poor performance of QTDT shown in our power simulations, future association studies should consider larger sample sizes, of the order of thousands of individuals, a much larger number of candidate gene loci and more powerful designs.
Accession codes
References
Abecasis GR, Cardon LR, Cookson WO (2000a). A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66: 279–292.
Abecasis GR, Cookson WO, Cardon LR (2000b). Pedigree tests of transmission disequilibrium. Eur J Hum Genet 8: 545–551.
Adams HD, Kolb TE (2004). Drought responses of conifers in ecotone forests of northern Arizona: tree ring growth and leaf δ13C. Oecologia 140: 217–225.
Agarwal PK, Agarwal P, Reddy MK, Sopory SK (2006). Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep 25: 1263–1274.
Allagulova ChR, Gimalov FR, Shakirova FM, Vakhitov VA (2003). The plant dehydrins: structure and putative functions. Biochemistry (Moscow) 68: 945–951.
Aranzana MJ, Kim S, Zhao K, Bakker E, Horton M, Jakob K et al. (2005). Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet 1: e60.
Asai T, Tena G, Plotnikova J, Willmann MR, Chiu WL, Gomez-Gomez L et al. (2002). MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 415: 977–983.
Baltunis BS, Huber DA, White TL, Goldfarb B, Stelzer HE (2007). Genetic analysis of early field growth of loblolly pine clones and seedlings from the same full-sib families. Can J For Res 37: 195–205.
Baltunis BS, Martin TA, Davis JM, Huber DA (2008). Inheritance of foliar stable carbon isotope discrimination and third-year height in Pinus taeda clones on contrasting sites in Florida and Georgia. Tree Genet Genomes (in press).
Brendel O, Pot D, Plomion C, Rozenberg P, Guehl JM (2002). Genetic parameters and QTL analysis of δ13C and ring width in maritime pine. Plant Cell Environ 25: 945–953.
Brown GR, Gill GP, Kuntz RJ, Langley CH, Neale DB (2004). Nucleotide variation and linkage disequilibrium in loblolly pine. Proc Natl Acad Sci USA 101: 15255–15260.
Cardon LR, Bell JI (2001). Association study designs for complex diseases. Nat Rev Genet 2: 91–99.
Cernusak LA, Aranda J, Marshall JD, Winter K (2007). Large variation in whole-plant water-use efficiency among tropical tree species. New Phytol 173: 294–305.
Chang S, Puryear JD, Dias MADL, Funkhouser EA, Newton RJ, Cairney (1996). Gene expression under water deficit in loblolly pine (Pinus taeda): isolation and characterization of cDNA clones. Physiol Plant 97: 139–148.
Close TJ (1997). Dehydrins: a commonalty in the response of plants to dehydration and low temperature. Physiol Plant 100: 291–296.
Cominelli E, Galbiati M, Vavasseur A, Conti L, Sala T, Vuylsteke M et al. (2005). A guard-cell-specific MYB transcription factor regulates stomatal movements and plant drought tolerance. Curr Biol 15: 1196–1200.
Dawson TE, Mambelli S, Plamboeck AH, Templer PH, Tu KP (2002). Stable isotopes in plant ecology. Annu Rev Ecol Syst 33: 507–559.
Dubos C, Plomion C (2003). Identification of water-deficit responsive genes in maritime pines (Pinus pinaster Ait.) roots. Plant Mol Biol 51: 249–262.
Ersoz ES (2006). Candidate gene-association mapping for dissecting fungal disease resistance in loblolly pine. PhD Dissertation, University of California, Davis, 293pp.
Ersoz ES, Yu J, Buckler ES (2007). Application of linkage disequilibrium and association mapping in crop plants. In: Varshney R, Tuberosa R (eds). Genomics-Assisted Crop Improvement. Springer: Netherlands, pp 97–119.
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci 5: 199–206.
Eveno E, Collada C, Guevara MA, Léger V, Soto A, Díaz L, Léger P, González-Martínez SC, Cervera MT, Plomion C, Garnier-Géré PH (2008). Contrasting patterns of selection at Pinus pinaster Ait. drought stress candidate genes as revealed by genetic differentiation analyses. Mol Biol Evol 25: 417–437.
Farquhar GD, Ehleringer JR, Hubick KT (1989). Carbon isotope discrimination and photosynthesis. Annu Rev Plant Physiol Plant Mol Biol 40: 503–537.
Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999). Combined linkage and association analysis for quantitative traits. Am J Hum Genet 64: 259–267.
González-Martínez SC, Ersoz E, Brown GR, Wheeler NC, Neale DB (2006). DNA sequence variation and selection of tag SNPs at candidate genes for drought-stress response in Pinus taeda L. Genetics 172: 1915–1926.
González-Martínez SC, Wheeler NC, Ersoz E, Nelson CD, Neale DB (2007). Association genetics in Pinus taeda L. I. Wood property traits. Genetics 175: 399–409.
Guillet-Claude C, Birolleau-Touchard C, Manicacci D, Rogowsky PM, Rigau J, Murigneux A et al. (2004). Nucleotide diversity of the ZmPox3 maize peroxidase gene: relationships between a MITE insertion in exon 2 and variation in forage maize digestibility. BMC Genet 5: 19.
Gupta PK, Rustgi S, Kulwal PL (2005). Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol Biol 57: 461–485.
Hirschhorn JN, Daly MJ (2005). Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6: 95–108.
Ingram J, Bartels D (1996). The molecular basis of dehydratation tolerance in plants. Annu Rev Plant Physiol Plant Mol Biol 47: 377–403.
Ismail AM, Hall AE, Close TJ (1999). Allelic variation of a dehydrin gene cosegregates with chilling tolerance during seedling emergence. Proc Natl Acad Sci USA 96: 13566–13570.
Karpinska B, Karlsson M, Schinkel H, Streller S, Süss KH, Melzer M et al. (2001). A novel superoxide-dismutase with a high isoelectric point in higher plants. Expression, regulation, and protein localization. Plant Physiol 126: 1668–1677.
Kiani SP, Grieu P, Maury P, Hewezi T, Gentzbittel L, Sarrafi A (2007). Genetic variability for physiological traits under drought conditions and differential expression of water stress-associated genes in sunflower (Helianthus annuus L). Theor Appl Genet 114: 193–207.
Kwok P-Y (2002). SNP genotyping with fluorescence polarization detection. Hum Mutat 19: 315–323.
Laird NM, Lange C (2006). Family-based designs in the age of large-scale gene-association studies. Nat Rev Genet 7: 385–394.
Lorenz WW, Sun F, Liang C, Kolychev D, Wang H, Zhao X et al. (2006). Water stress-responsive genes in loblolly pine (Pinus taeda) roots identified by analyses of expressed sequence tag libraries. Tree Physiol 26: 1–16.
Luu DT, Maurel M (2005). Aquaporins in the challenging environment: molecular gears for adjusting plant water status. Plant Cell Environ 28: 85–96.
Martínez-Vilalta J, Sala A, Piñol J (2004). The hydraulic architecture of Pinaceae—a review. Plant Ecology 171: 3–13.
Mckay JK, Richards JH, Mitchell-Olds T (2003). Genetics of drought adaptation in Arabidopsis thaliana: I. Pleiotropy contributes to genetic correlations among ecological traits. Mol Ecol 12: 1137–1151.
Monclus R, Dreyer E, Villar M, Delmotte FM, Delay D, Petit JM et al. (2006). Impact of drought on productivity and water use efficiency in 29 genotypes of Populus deltoides x Populus nigra. New Phytol 169: 765–777.
Monks SA, Kaplan NL, Weir BS (1998). A comparative study of sibship tests of linkage and/or association. Am J Hum Genet 63: 1507–1516.
Mousavi A, Hotta Y (2005). Glycine-rich proteins: a class of novel proteins. Appl Biochem Biotechnol 120: 169–174.
Neale DB, Savolainen O (2004). Association genetics of complex traits in conifers. Trends Plant Sci 9: 325–330.
Newton RJ, Funkhouser EA, Fong F, Tauer CG (1991). Molecular and physiological genetics of drought tolerance in forest species. For Ecol Manag 43: 225–250.
Nguyen-Queyrens A, Bouchet-Lannat F (2003). Osmotic adjustment in three-year-old seedlings of five provenances of maritime pine (Pinus pinaster) in response to drought. Tree Physiol 23: 397–404.
Oliveras I, Martínez-Vilalta J, Jimenez-Ortiz T, Lledo MJ, Escarré A, Piñol J (2003). Hydraulic properties of Pinus halepensis, Pinus pinea and Tetraclinis articulata in a dune ecosystem of Eastern Spain. Plant Ecology 169: 131–141.
Peter G, Neale DB (2004). Molecular basis for the evolution of xylem lignification. Curr Opin Plant Biol 7: 737–742.
Reddy AR, Chaitanyaa KV, Vivekanandanb M (2004). Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J Plant Physiol 161: 1189–1202.
Richard S, Morency MJ, Drevet C, Jouanin L, Séguin A (2000). Isolation and characterization of a dehydrin gene from white spruce induced upon wounding, drought and cold stresses. Plant Mol Biol 43: 1–10.
Riera M, Valon C, Fenzi F, Giraudat J, Leung J (2005). The genetics of adaptive responses to drought stress: abscisic acid-dependent and abscisic acid-independent signalling components. Physiol Plant 123: 111–119.
Robatzek S, Somssich IE (2002). Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev 16: 1139–1149.
Rorat T (2006). Pland dehydrins—tissue location, structure and function. Cell Mol Biol Lett 11: 536–556.
Sathyan P, Newton RJ, Loopstra CA (2005). Genes induced by WDS are differentially expressed in two populations of aleppo pine (Pinus halepensis). Tree Genet Genomes 1: 166–173.
Schmidtling RC (2001). Southern pine seed sources. USDA, 6TR SRS-44, Asheville, NC.
Shinozaki K, Yamaguchi-Shinozaki K (2007). Gene networks involved in drought stress response and tolerance. J Exp Bot 58: 221–227.
Spielman RS, McGinnis RE, Ewens WJ (1993). Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52: 506–516.
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler IV ES (2001). Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28: 286–289.
Thumma BR, Nolan MF, Evans R, Moran GF (2005). Polymorphisms in Cinnamoyl CoA Reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171: 1257–1265.
Tjoelker MG, Oleksyn J, Reich PB (1998). Seedlings of five boreal tree species differ in acclimation of net photosynthesis to elevated CO2 and temperature. Tree Physiol 18: 715–726.
Ülker B, Somssich IE (2004). WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol 7: 491–498.
Wang W, Vinocur B, Altman A (2003). Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218: 1–14.
Warren CR, McGrath JF, Adams MA (2001). Water availability and carbon isotope discrimination in conifers. Oecologia 127: 476–486.
Wilson LM, Whitt SR, Ibáñez AM, Rocheford TR, Goodman MM, Buckler IV ES (2004). Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16: 2719–2733.
Yu J, Pressoir G, Briggs WH, Vroh-Bi I, Yamasaki M, Doebley JF et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38: 203–208.
Zhang Y, Li J, Yu F, Cong L, Wang L, Burkard G et al. (2006). Cloning and expression analysis of SKn-type dehydrin gene from bean in response to heavy metals. Mol Biotechnol 32: 205–217.
Zhang Y, Wang Z, Xu J (2007). Molecular mechanism of dehydrin in response to environmental stress in plant. Prog Nat Sci 17: 237–246.
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C et al. (2007). An Arabidopsis example of association mapping in structured samples. PLoS Genet 3: e4.
Acknowledgements
We thank Ricardo Alía for valuable comments and discussions and PC Grant for English language suggestions. Robert J Kuntz and Julie Beal provided technical assistance in the laboratory. The study of SC González-Martínez was supported by a Fulbright/MECD scholarship at the University of California (Davis) and the ‘Ramón y Cajal’ fellowship RC02-2941 (Spanish Ministry of Science). This research was supported by the ADEPT (Allele Discovery for Genes Controlling Economic Traits in Loblolly Pine) project funded in the framework of the Initiative for Future Agriculture and Food Systems (USDA, USA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
González-Martínez, S., Huber, D., Ersoz, E. et al. Association genetics in Pinus taeda L. II. Carbon isotope discrimination. Heredity 101, 19–26 (2008). https://doi.org/10.1038/hdy.2008.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2008.21
Keywords
This article is cited by
-
Genomic selection: a revolutionary approach for forest tree improvement in the wake of climate change
Euphytica (2024)
-
Genotyping SNPs in lignin biosynthesis gene (CAD1) and transcription factors (MYB1 and MYB2) exhibits association with wood density in teak (Tectona grandis L.f.)
Molecular Biology Reports (2024)
-
Genome-wide association studies: an intuitive solution for SNP identification and gene mapping in trees
Functional & Integrative Genomics (2023)
-
Genetic variation in carbon isotope discrimination-based water use efficiency of teak (Tectona grandis L. f.) and its association with growth and wood quality traits
Trees (2021)
-
Marker associations for fusiform rust resistance in a clonal population of loblolly pine (Pinus taeda, L.)
Tree Genetics & Genomes (2020)
