
Abstract
Purpose: To determine the size and parental origin of the deletion in individuals with 18p− syndrome.
Methods: Molecular and fluorescence in situ hybridization analyses of the pericentromeric region of chromosome 18 were performed on genomic DNA and chromosomes from study participants.
Results: The majority of the breakpoints were located between markers D18S852 on 18p and D18S1149 on 18q, a distance of approximately 4 Mb. The parental origin of these deletions appears to be equally distributed, half maternally derived and half paternally derived.
Conclusion: The distributions of both the size and parental origin of the 18p deletions support the presence of a breakpoint cluster in the 18p− syndrome.
Similar content being viewed by others


Main
When it was first described in 1963, the 18p− syndrome was the only autosomal deletion syndrome then known to be compatible with long-term survival.1 Reported clinical features include speech delay, growth hormone deficiency,2 cardiac anomalies,3 ptosis, and holoprosencephaly.4 Mental retardation has also been associated with this syndrome, but this phenotype has wide variation, ranging from severe mental retardation to borderline normal intelligence.5,6
We are interested in understanding the molecular basis of the clinical characteristics in this syndrome. Detailed molecular analyses were used to ascertain the size and parental origin of the deletion in each subject. Determining the exact size of the deletion is necessary for identifying critical regions and candidate genes that may contribute to a phenotype. Identification of critical regions, using molecular and cytogenetic methods, has already been done for other aneusomies such as Turner syndrome,7 Down syndrome,8 cri-du-chat (5p-) syndrome,9 and 18q− syndrome.10,11
It is important to know the parental origin of the chromosome with the deletion for two reasons. First, there can be differences in the origin of the deletion, dependent on the sex of the parent. Parental origin biases in mutation rates were reviewed by Chandley.12 Chromosome abnormalities in particular have exhibited parent-of-origin biases. Abnormalities resulting from nondisjunction, such as trisomies 18 and 21, have a maternal parent of origin tendency,13–15 whereas structural rearrangements such as terminal deletions have an increased paternal origin bias.16 Developmental differences between oogenesis and spermatogenesis may give rise to a parent of origin bias in individuals with chromosomal abnormalities. The paternal bias in structural rearrangements is thought to be due to the greater number of cell divisions during male meiosis, as well as minimal repair capacity in sperm.12
Second, the parental origin of a deleted chromosome can also have an effect on phenotypic outcome, a process known as imprinting. The most commonly cited example is the microdeletion 15q11-q13. If the microdeletion is maternally derived or there is paternal uniparental disomy of chromosome 15, the child will have Angelman syndrome. If the 15q11-q13 microdeletion is paternally derived or there is maternal uniparental disomy, the child will have Prader-Willi syndrome.17 As this project is part of a larger genotype/phenotype correlation study of the 18p− syndrome, we wanted to determine the parental origin of the deletion to look for parental age and imprinting effects.
Our results describe the molecular analyses of the largest group of 18p− patients in a single study. We demonstrate the presence of a possible breakpoint cluster on the short arm of chromosome 18 and the absence of a parent-of-origin bias for terminal de novo deletions, regardless of where on the chromosome the deletion occurs. We also show a statistically significant difference in average maternal age between those deletions in the putative breakpoint cluster and those that are not in the breakpoint cluster region.
Subjects and Methods
Patients
Twenty-five subjects with deletions of 18p were recruited from The Chromosome 18 Registry and Research Society. Written informed consent was obtained for all subjects under an institutional review board-approved protocol. The 25 unrelated subjects provided medical records and blood samples. If cytogenetic reports were not available, the proband and parental karyotypes were repeated by our laboratory with G-banding analysis of at least a 550-band level. All individuals with 18p deletions that were the result of inherited unbalanced translocations or other rearrangements were excluded from the analysis. Only nonmosaic patients with de novo deletions of 18p were included in the study.
Microsatellite analysis
Genomic DNA was isolated from whole blood using the PUREGENE DNA Isolation kit (Gentra Systems; Minneapolis, MN). Molecular analysis of the deletions was completed by PCR with polymorphic microsatellite primers. All markers used were from Généthon18 with the exception of D18S852 (Cooperative Human Linkage Center) and D18S40.19 Initially, the microsatellite markers chosen were evenly distributed on 18p. After the apparent breakpoint cluster was identified, additional microsatellite markers were selected from the pericentromeric region to further define the location of the breakage site.
Reaction conditions were 50 ng template DNA, 50 ng of each primer, 1.5 mM MgCl2, 50 mM KCl, 200 μM dNTPs, and 1 U of Taq polymerase (Applied Biosystems, Inc., Foster City, CA). The forward primer was end-labeled with 0.5 μCi [γ-32P]ATP using 0.4 U of T4 polynucleotide kinase. An initial denaturation step of 5 minutes at 94°C was followed by 35 cycles of 1 minute at 94°C, 1 minute at an average annealing temperature of 55°C, and 1 minute at 72°C, with a final extension at 72°C for 10 minutes. Annealing temperatures for each set of primers ranged from 50 to 65°C depending on the GC content of the primers. Products were electrophoresed on a 7% acrylamide gel.
Fluorescence in situ hybridization
In cases where markers in the pericentromeric region were uninformative, fluorescence in situ hybridization (FISH) was performed to determine the approximate boundaries of the breakpoint cluster. An alpha satellite probe (Vysis, Inc., Downers Grove, IL) was used to confirm the presence of the centromere when D18S1149, the closest marker to the centromere on 18q, was uninformative. To define the breakpoint on the short arm of chromosome 18 when both D18S40 and D18S852 were uninformative, a BAC genomic clone containing the marker D18S852 was identified by electronic PCR in NCBI (http://www.ncbi.nlm.nih.gov) and ordered from BACPAC Resources (Buffalo, NY). DNA from this clone, RP11–808F20, was isolated with the Qiagen plasmid maxi kit (Valencia, CA) and also used as a probe for FISH.
Fresh metaphase spreads were baked for 4 hours at 65°C. The chromosomes were denatured for 2 minutes in 70% formaldehyde, 2× SSC (pH 7.0), dehydrated through an ethanol series, and dried. The probes were labeled with biotin-14-dATP by nick translation using a BioNick kit (Gibco BRL, Rockville, MD). The hybridization solution for each slide contained 40 ng labeled probe and 10 μg human Cot-1 DNA. After the DNA was denatured for 5 minutes at 70°C, the probes were added to the slides and were incubated overnight at 37°C. Next day, the probes were fluorescently tagged using avidin-fluorescein isothiocyanate (avidin-FITC; Vector Laboratories, Burlingame, CA). Signal amplification was achieved using biotin-labeled antiavidin (Vector Laboratories), followed by avidin-FITC. The chromosomes were counterstained with DAPI and visualized on a Zeiss Axioscope microscope. Digital images were captured using an imaging system and Provision Software from Applied Imaging (Santa Clara, CA).
Statistical methods
One-way analysis of variance was used to compare mean differences in parental age according to parental origin and deletion subgroups of interest, followed by a Tukey studentized range posthoc test to adjust for multiple comparisons20 (Table 1).
RESULTS
Deletion size and distribution
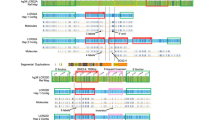
Figure 1 shows the breakpoint estimation using PCR based polymorphic markers. Of the total study population, 18 subjects had breakpoints between markers D18S852 and D18S1149 (72%), a map distance of 4 Mb, creating an apparent breakpoint cluster in the pericentromeric region on 18p. Current map distances on Figure 1 were obtained from the April 1, 2001, freeze of the sequence data obtained from The Human Genome Project Working Draft Web site at the University of California at Santa Cruz (http://genome.ucsc.edu).

Molecular analysis of subjects with chromosome 18p deletions using polymorphic markers. Data from each participant is shown in a column under their study number. Closed circles indicate that the subject has two alleles for that marker. Open circles indicate that the subject has a single allele. Genotyping performed by fluorescence in situ hybridization is indicated by open or closed triangles. When the marker was uninformative, no mark is shown. The minimal region of the retained part of the chromosome is enclosed in a box.
Only seven subjects had breakpoints that were not located in the pericentromeric region of chromosome 18p. Patient 29 had a breakpoint between markers D18S458 and D18S1132, a distance of approximately 1 Mb, Patient 18 between markers D18S852 and D18S464 (5 Mb), Patient 17 between markers D18S453 and D18S976 (8 Mb), Patient 20 between markers D18S458 and D18S464 (4.5 Mb), Patient 22 between markers D18S464 and D18S1154 (7 Mb), and Patient 24 between markers D18S71 and D18S1116 (1 Mb). There was no specific pattern to the distribution of breakpoints in these individuals.
Parental origin
The maternal/paternal origin ratio for the entire group was 13:12, approximately 50:50. To determine whether parental origin differences were related to breakpoint location, parental origin ratios were compared between two groups: those with breakpoints in the pericentromeric region of 18 versus those not in the pericentromeric region. No significant difference between the groups was evident. In the first category, 10 deletions were of maternal origin and 8 of paternal origin; for those not in the pericentromeric group, 3 were maternal and 4 paternal.
To determine whether advanced parental age might be a causative factor in the 18p− syndrome, the mean differences in parental age at conception were assessed according to deletion and parental origin subgroups using one-way analysis of variance followed by a Tukey studentized range posthoc test to adjust for multiple comparisons20 (Table 1). Comparison of mean maternal age scores between breakpoint cluster (31.6 years) and nonbreakpoint cluster (27.0 years) subgroups exhibited a statistically significant difference (P = 0.02). In contrast, comparison of mean paternal age scores between breakpoint cluster (31.7 years) and nonbreakpoint cluster (29.7 years) subgroups did not exhibit a statistically significant difference (P = 0.14). Examination of the post hoc analyses indicates that no statistically significant differences in either maternal or paternal age (P < 0.05) were exhibited according to parental origin subgroup within either the breakpoint cluster or the nonbreakpoint cluster subgroups.
DISCUSSION
The parental origin of a de novo chromosomal deletion is usually paternal. Examples of deletion syndromes with a strong paternal origin bias are Wolf-Hirschorn syndrome (4p-),21 cri-du-chat syndrome (5p-),9,22 and the 18q− syndrome.16 One exception is 11q−, otherwise known as Jacobsen syndrome. Similar to our findings in the 18p− syndrome, the parental origin ratios are approximately 50:50 and there is also a breakpoint cluster present.23 The hypothesis for the formation of the breakpoint cluster in Jacobsen syndrome is the expression of a folate-sensitive fragile site, FRA11B, located in 11q23.3.24 Michaelis et al.25 documented the relationship in 11q− between parental origin of the deletion and the location of the breakpoint. Most maternally derived deletions are caused by breakage at FRA11B, whereas most paternally derived deletions occur distal to the fragile site.
Based on these similarities, we first hypothesized that a fragile site might be located at or near 18p11.1. However, there is no documented fragile site on 18p, despite our findings of an apparent breakpoint cluster. In addition, unlike the 11q− syndrome, the evenly split parental origin ratio remained the same regardless of whether the chromosome break occurred at the putative breakpoint cluster or at a more distal location on the chromosome. No bias toward paternal origin was detected in those subjects that had breakpoints that were not in the pericentromeric region. However, our small sample size (N = 7) prevents the formation of any definitive conclusions.
Although there is no known fragile site on 18p near the centromere, recent studies emphasize the importance of normally methylated pericentromeric DNA as a prerequisite for proper chromosome segregation and stabilization. Undermethylated pericentromeric satellite DNA and chromosomal abnormalities are found in the lymphocytes of patients with ICF, a genetic disease diagnosed by the combination of immunodeficiency, centromeric instability, and facial anomalies.26 DNA hypomethylation is also strongly correlated with chromosomal instability in human cancers.27 Certain polymorphisms in genes involved in folate metabolism and undermethylation of pericentromeric heterochromatin have been implicated as additional maternal risk factors for having a child with Down syndrome.28 In addition, the fragile site expressed in Jacobsen syndrome is folate-dependent.24 Folate-dependent fragile sites are caused by the expansion of CCG-repeats and the hypermethylation of adjacent CpG islands. Deletion breakpoints of paternal origin at more distal locations in Jacobsen syndrome have also been shown to contain CCG-repeats.29 Given these data, abnormal methylation of pericentromeric DNA in oogenesis or spermatogenesis, followed by breakage and the de novo addition or capture of a telomere,30 could explain the occurrence of the breakpoint cluster on 18p. Our statistical analysis of parental ages suggests that advanced maternal age may also be a contributing factor to the occurrence of pericentromeric 18p deletions.
The common distribution of breakpoints and the approximately equal parental origin ratios in our study population are also consistent with a mechanism of unequal crossing over between homologous chromosomes in meiosis I, resulting in monosomy 18p.31 Unequal recombination is the model proposed for some genetic syndromes caused by interstitial deletions, such as Williams syndrome32 and DiGeorge syndrome/VCFS.33 In most interstitial deletion syndromes, there is a common breakpoint region, a common deletion size, and an equal probability that the deletion is derived from either the paternal or the maternal chromosome.31 Interestingly, the marker order on the short arm of chromosome 18 in the genetic linkage map of the baboon (Papio hamadryas) genome is inverted in comparison with that of the human linkage map,34 suggesting that sequences in the pericentromeric region of 18p may be able to either serve as telomeres or as templates for telomerase, or have done so at some point in their evolutionary history. Whether any of the cytogenetically defined terminal deletions in our study population are cryptic rearrangements or actually interstitial deletions that have retained the original telomeres, will be addressed in future experiments.
As a result of the current study, we obtained data that clearly demonstrates the presence of a breakpoint cluster on 18p. Although we realize that some designated breakpoint clusters in other syndromes encompass smaller regions than 4 Mb, the distance could be narrowed when the fine mapping of this region is completed. The common breakpoint distribution that we found in our study population is in sharp contrast to that of other terminal deletion syndromes, including monosomy 1p3635 and particularly the 18q− syndrome,16 in which every individual appears to have a unique breakpoint. The parental origin of the 18p deletions appears to be equally distributed, half maternally derived and half paternally derived. Interestingly, in studies of other terminal deletion syndromes, all but one describes preferential loss of the paternal chromosomes.9,16,21,22 The one exception to this preferential distribution is the 11q− syndrome, also known as Jacobsen syndrome, which showed equal maternal and paternal distribution of chromosomal loss at a fragile site.23 Thus, both the breakpoint cluster and the parent-of-origin distribution support the presence of a hot spot for chromosomal breakage in the 18p− syndrome. Statistical analyses of average parental ages document an advanced maternal age bias for deletion breakpoints occurring in the pericentromeric region of 18p.
Further studies are needed to map the breakpoint region(s) in greater detail and to identify possible mechanisms for breakage. Also, clinical studies of the patients in this study have been initiated for the purpose of genotype/phenotype mapping. The detailed molecular analyses already performed should assist in elucidating the pathogenesis of the clinical manifestations and identifying critical regions and genes that are responsible for the 18p− phenotype.
References
De Grouchy J, Lamy M, Thieffry S, Arthuis M, Salmon CH . Dysmorphie complexe Avec oligophrénie: deletion des bras courts d'un chromosome 17–18. C R Acad Sci 1963; 256: 1028.
Schober E, Scheibenreiter S, Frisch H . 18p monosomy with GH-deficiency and empty sella: good response to GH-treatment. Clin Genet 1995; 47: 254–256.
Pearl W . Heart disease associated with deletion of the short arm of chromosome 18. Pediatr Cardiol 1989; 10: 174–176.
Faust J, Habedank M, Nieuwenhuijsen C . The 18p– syndrome: report of four cases. Eur J Pediatr 1976; 123: 59–66.
Horsley SW, Knight SJL, Nixon J, Huson S, Fitchett M, Boone RA, Hilton-Jones D, Flint J, Kearney L . Del(18p) shown to be a cryptic translocation using a multiprobe FISH assay for subtelomeric chromosome rearrangements. J Med Genet 1998; 35: 722–726.
Thompson RW, Peters JE, Smith SD . Intellectual, behavioral, and linguistic characteristics of three children with 18p– syndrome. J Dev Behav Pediatr 1986; 7: 1–7.
Zinn AR, Tonk VS, Chen Z, Flejter WL, Gardner HA, Guerra R, Kushner H, Schwartz S, Sybert VP, Van Dyke DL, Ross JL . Evidence for a Turner Syndrome locus or loci at Xp11.2-p22.1. Am J Hum Genet 1998; 63: 1757–1766.
Epstein EJ, Korenberg JR, Annéren G, Antonarakis SE, Aymé S, Courchesne E, Epstein LB, Fowler A, Groner Y, Huret JL, Kemper TL, Lott IT, Lubin BH, Magenis E, Opitz JM, Patterson D, Priest JH, Pueschel SM, Rapoport SI, Sinet PM, Tanzi RE, de la Cruz F . Protocols to establish genotype-phenotype correlations in Down Syndrome. Am J Hum Genet 1991; 49: 207–235.
Church DM, Bengtsson U, Nielson KV, Wasmuth JJ, Niebuhr E . Molecular definition of deletions of different segments of distal 5p that result in distinct phenotypic features. Am J Hum Genet 1995; 56: 1162–1172.
Cody JD, Hale DE, Brkanac Z, Kaye CI, Leach RJ . Growth hormone insufficiency associated with haploinsufficiency at 18q23. Am J Med Genet 1997b; 71: 420–425.
Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, Hoffman DS, Muller S, Schaub RL, Leach RJ, Kaye CI . Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet 1999; 85: 455–462.
Chandley AC . On the parental origin of de novo mutation in man. J Med Genet 1991; 28: 217–223.
Bricarelli FD, Pierluigi M, Grasso M, Strigini P, Perroni L . Origin of extra chromosome 21 in 343 families: cytogenetic and molecular approaches. Am J Med Genet 1990; 7: 129–132.
Nothen MM, Eggerman T, Erdmann J, Eiben B, Hofmann D, Propping P, Schwanitz G . Retrospective study of the parental origin of the extra chromosome in trisomy 18 (Edwards syndrome). Hum Genet 1993; 92: 347–349.
Ya-gang X, Robinson WP, Spiegel R, Binkert F, Ruefenacht U, Schinzel AA . Parental origin of the supernumerary chromosome in trisomy 18. Clin Genet 1993; 44: 57–61.
Cody JD, Pierce JF, Brkanac Z, Plaetke R, Ghidoni P, Kaye CI, Leach RJ . Preferential loss of the paternal alleles in the 18q– syndrome. Am J Med Genet 1997; 69: 280–286.
Glenn CC, Driscoll DJ, Yang TP, Nicholls RD . Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod 1997; 3: 321–322.
Gyapay G, Morissette J, Vignal A, Dib C, Fizames C, Millasseau P, Marc S, Bernardi G, Lathrop M, Weissenbach J . The 1993–94 Généthon human genetic linkage map. Nat Genet 1994; 7: 246–339.
Straub RE, Speer MC, Luo Y, Rojas K, Overhauser J, Ott J, Gilliam TC . A microsatellite linkage map of human chromosome 18. Genomics 1993; 15: 48–56.
Cody R, Smith J . Applied statistics and the SAS programming language. New Jersey: Prentice Hall, 1997.
Tupler R, Bortotto L, Buhler EM, Alkan M, Malik MJ, Bosch-AlJadooa N, Memo L, Maraschio P . Paternal origin of the de novo deleted chromosome 4 in Wolf-Hirschorn syndrome. J Med Genet 1992; 29: 53–55.
Overhauser J, McMahon J, Oberlender S, Carlin ME, Niebuhr E, Wasmuth JJ, Lee-Chen J . Parental origin of the Chromosome 5 deletions in the Cri-du-Chat syndrome. Am J Med Genet 1990; 37: 83–86.
Penny LA, Dell'Aquila M, Jones MC, Bergoffen J, Cunniff C, Fryns JP, Grace E, Graham JM, Kousseff B, Mattina T, Syme J, Voullaire L, Zelante L, Zenger-Hain J, Jones OW, Evans GA . Clinical and molecular characterization of patients with distal 11q deletions. Am J Hum Genet 1995; 56: 676–683.
Jones C, Slijepcevic P, Marsh S, Baker E, Langdon WY, Richards RI, Tunnacliffe A . Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum Mol Genet 1994; 3: 2123–2130.
Michaelis RC, Velagaleti GV, Jones C, Pivnick EK, Phelan MC, Boyd E, Tarleton J, Wilroy RS, Tunnacliffe A, Tharapel AT . Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. Am J Med Genet 1998; 76: 222–228.
Miniou P, Jeanpierre M, Bourchis D, Barbosa ACC, Blanquet V, Viegas-Pequignot E . α-satellite DNA. methylation in normal individuals and in ICF patients: heterogeneous methylation of constitutive heterochromatin in adult and fetal tissues. Hum Genet 1997; 99: 738–745.
Vilain A, Vogt N, Dutrillaux B, Malfoy B . DNA methylation and chromosome instability in breast cancer cell lines. FEBS Lett 1999; 460: 231–234.
Hobbs CA, Sherman SL, Yi P, Hopkins SD, Torfs CP, Hine RJ, Pogribna M, Rozen R, James SJ . Polymorphisms in genes involved in folate metabolism as maternal risk factors for Down syndrome. Am J Hum Genet 2000; 67: 623–630.
Jones C, Müllenbach R, Grossfeld P, Auer R, Favier R, Chien K, James M, Tunnacliffe A, Cotter F . Co-localisation of CCG repeats and chromosome deletion breakpoints in Jacobsen syndrome: evidence for a common mechanism of chromosome breakage. Hum Mol Genet 2000; 9: 1201–1208.
Meltzer PS, Guan X-Y, Trent JM . Telomere capture stabilizes chromosome breakage. Nat Genet 1993; 4: 252–255.
Shaffer LG, Lupski JR . Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet 2000; 34: 297–329.
Nickerson E, Greenberg F, Keating MT, McCaskill C, Shaffer LG . Deletions of the elastin gene at 7q11.23 occur in approximately 90 of patients with Williams syndrome. Am J Hum Genet 1995; 56: 1156–1161.
Morrow B, Goldberg R, Carlson C, Das Gupta R, Sirotkin H, Collins J, Dunham I, O'Donnell H, Scambler P, Shprintzen R, Kucherlapati R . Molecular definition of the 22q11 deletions in velo-cardio-facial syndrome. Am J Hum Genet 1995; 56: 1391–1403.
Rogers J, Mahaney MC, Witte SM, Nair S, Newman D, Wedel S, Rodriguez LA, Rice KS, Slifer SH, Perelygin A, Slifer M, Palladino-Negro P, Newman T, Chambers K, Joslyn G, Parry P, Morin PA . A genetic linkage map of the baboon(Papio hamadryas)genome based on human microsatellite polymorphisms. Genomics 2000; 67: 237–247.
Shapira SK, McCaskill C, Northrup H, Spikes AS, Elder FF, Sutton VR, Korenberg JR, Greenberg F, Shaffer LG . Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet 1997; 61: 642–650.
Acknowledgements
This research was supported by a grant from the Chromosome 18 Registry and Research Society and by the San Antonio Cancer Institute (grant P30 CA 54174). We are grateful to the subjects and their families for their participation in the study. We also thank the reviewers of this manuscript for their valuable and constructive comments.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Schaub, R., Reveles, X., Baillargeon, J. et al. Molecular characterization of 18p deletions: Evidence for a breakpoint cluster. Genet Med 4, 15–19 (2002). https://doi.org/10.1097/00125817-200201000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200201000-00003
Keywords
This article is cited by
-
Adults with Chromosome 18 Abnormalities
Journal of Genetic Counseling (2015)
-
Monosomy 18p
Orphanet Journal of Rare Diseases (2008)
-
Towards mapping phenotypical traits in 18p− syndrome by array-based comparative genomic hybridisation and fluorescent in situ hybridisation
European Journal of Human Genetics (2007)
-
Familial deletion 18p syndrome: case report
BMC Medical Genetics (2006)
