
Abstract
The branchio–oto–renal (BOR) syndrome is an autosomal-dominant disorder characterized by hearing loss, branchial and renal anomalies. BOR is genetically heterogeneous and caused by mutations in EYA1 (8q13.3), SIX1 (14q23.1), SIX5 (19q13.3) and in an unidentified gene on 1q31. We examined six Danish families with BOR syndrome by assessing linkage to BOR loci, by performing EYA1 multiplex ligation-dependent probe amplification (MLPA) analysis for deletions and duplications and by sequencing of EYA1, SIX1 and SIX5. We identified four EYA1 mutations (c.920delG, IVS10−1G>A, IVS12+4A>G and p.Y591X) and one SIX1 mutation (p.W122R), providing a molecular diagnosis in five out of the six families (83%). The present, yet preliminary, observation that renal and temporal bone malformations are less frequent in SIX1-related disease suggests a slightly different clinical profile compared to EYA1-related disease. Unidentified mutations impairing mRNA expression or further genetic heterogeneity may explain the lack of mutation finding in one family despite LOD score indications of EYA1 involvement.
Similar content being viewed by others

Introduction
The branchio–oto–renal (BOR) syndrome (OMIM 113650) is an autosomal-dominant developmental disorder, associated with hearing loss, branchial arch and renal anomalies.1 The highly variable phenotype in patients with BOR syndrome comprises hearing loss (93%), preauricular pits (82%), renal anomalies (67%), branchial fistulae (49%), deformed pinnae (36%) and external auditory canal stenosis (29%).2 Less frequent findings include preauricular tags (13%) and lacrimal duct aplasia (11%).2 A set of diagnostic criteria was suggested by Chang et al.3 Based on clinical presentation, the prevalence of BOR was estimated to be 1:40 000, and to affect approximately 2% of profoundly deaf children.4
Hearing loss is a crucial finding, presenting as sensorineural, conductive or mixed type with about equal distribution of the three types of hearing loss. The hearing impairment may be progressive and fluctuating.5 Temporal bone abnormalities include enlarged vestibular aqueduct (EVA), hypoplastic cochleae and hypoplastic semicircular canals,6, 7, 8, 9 but it should be mentioned that molecular diagnosis was absent in these early reports. In one study, applying a standardized set of measurements, 76.9% of patients had either unilateral or bilateral CT abnormalities.7 A recent study reports EVA in a patient with a SIX1 mutation.10
BOR is genetically heterogeneous and is associated with mutations in EYA1 on chromosome 8q13.3, SIX1 on 14q23.1, and a third BOR locus, BOR2, on 1q31.11, 12, 13 After submission of this article, the SIX1 homologue SIX5 (19q13.3) has been found to be mutated in BOR patients.14 At least 116 different EYA1 mutations and rearrangements have been reported (isoform B).3, 15, 16 By contrast, only few mutations have been reported in the SIX genes. Thus, so far, five unrelated families have been published with three different mutations in SIX1,10, 17 and five BOR individuals have been identified with four different SIX5 missense mutations.14
Alternative splicing of EYA1 produces four transcript variants, expressing three different isoforms with the longest isoform having 18 exons.3 Eya family members (Eya1–4) are defined by a conserved ∼275 amino-acid C-terminal domain, referred to as the Eya domain and which possesses protein phosphatase activity.18 Eya proteins, including Eya1, function as co-activators in transcriptional regulation by Dach proteins and Six proteins (including Six1 and Six5) during mammalian organogenesis.17, 18, 19 Eya has no apparent DNA-binding ability and is translocated from the cytoplasm to the nucleus by Six and serves as a co-activator of Six in the regulation of downstream genes.19, 20 Both Eya and Dach may be required for Six-mediated gene activation.19 The recently discovered protein phosphatase function of the Eya family is thought to switch Six-Dach from a repressor to an activator of transcription via dephosphorylation.18, 19
The aim of this study was to identify the disease causing mutations in six Danish families diagnosed with BOR, of which five have been published with clinical description previously. Family information was updated, and linkage analysis to EYA1, SIX1 and BOR2 locus, respectively, was performed, followed by EYA1 multiplex ligation-dependent probe amplification (MLPA) analysis, and sequencing of EYA1, SIX1 and SIX5.
Materials and methods
Patients and families
We used the clinical criteria suggested by Chang et al3 in which an individual must fulfil one of the following criteria to be classified as affected with BOR: (A) fulfil at least three major criteria (branchial anomalies, deafness, pre-auricular pits, renal anomalies), or (B) fulfil two major criteria and at least two minor criteria (external, middle or inner ear anomalies, pre-auricular tags, and possibly also have other features such as facial asymmetry or palate abnormalities) or (C) fulfil one major criteria and have an affected first-degree relative meeting the criteria for BOR.3 Six Danish families diagnosed with BOR participated in the study (Figure 1). Family 1,21 3,22 4,22 56 and 622 have been described previously, whereas family 2 is new. The clinical data were compiled from previous publications and from available medical records from different hospitals. Venous blood was drawn from all available patients and relatives. The study was approved by the Danish Research Ethical Committee (reference numbers KF 01–234/02 and KF 01–108/03).


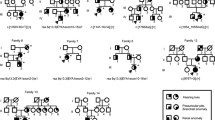
Pedigrees of the Danish BOR families 1–6. Probands are indicated by arrows. Asterisks (*) indicate individuals who were include in linkage analysis. (a) Pedigree of family 1 with haplotypes for EYA1 on chromosome 8 with markers D8S381, D8S1795 and D8S279. The c.1773C>G mutation segregated with BOR in the family. Individual IV-5 turned out to be a phenocopy. (b) Pedigree showing family 2. The c.364T>A mutation in SIX1 segregated with BOR in the family. Individuals V-10, VI:7, VI:8 and VI:9 were not included in linkage analysis due to late arrival of the blood samples. (c) Pedigree showing family 3. The mutation, ISV10−1G>A (c.1141−1G>A), in EYA1 segregated with BOR in the family. (d) Pedigree showing family 6. The c.917delG mutation in EYA1 segregated with BOR in the family. (e) Pedigree showing family 4 with haplotypes for EYA1 on chromosome 8 with marker D8S381, D8S1795 and D8S279. No mutation was identified in this family. (f) Pedigree showing family 5. The mutation, ISV12+4A>G (c.1360A>G), in EYA1 segregated with BOR in the family. Individuals V:14 and VI:13 are phenocopies.
DNA isolation and chromosome analysis
DNA was extracted from peripheral blood samples using standard methods. Chromosome analysis of cultivated leucocytes in a patient from family 4 (individual III:5) followed standard procedures for karyotyping.
Marker analysis
The following markers were chosen to cover the three BOR loci: D8S381, D8S1795 and D8S279 for linkage to EYA1; D1S2757 for linkage to the BOR2 locus; and D14S980 and D14S290 for linkage to SIX1. In one family, additional markers were used to saturate the BOR2 region: D1S2640, D1S238, D1S2757, D1S1660, D1S2654, D1S2872 and D1S249. The forward primers for the markers were labelled using γ-[33P]-dATP (Hartmann Analytic, Helsingborg, Sweden) and T4-DNA-polynucleotide kinase (Fermentas, Braunschweig, Germany) according to the manufacturer's protocol. PCR was carried out using 50 ng DNA and Taq-polymerase (New England Biolabs, USA), and PCR products were separated by 5% acrylamide gel electrophoresis and exposed to X-ray films.
Linkage analysis
The two-point LOD scores and multipoint LOD scores were calculated using the computer program MLINK and LINKMAP routines of the FASTLINK software version 2.2. The multipoint LOD score was calculated based on microsatellite markers as reported in the GDB database (www.gdb.org). The LOD scores were calculated by using a dominant model with complete penetrance (100%).
MLPA analysis
MLPA analysis detects deletions or duplications of one or more exons, but not inversions. The MLPA-EYA1 kit (P153) was designed by MRC-Holland, Amsterdam, The Netherlands, upon the authors’ suggestion. The EYA1 probe mix contains 17 probes for EYA1 exons and 14 control probes. To test the MLPA-EYA1 kit, a DNA sample carrying an EYA1 deletion previously identified by other methods was reanalysed using the kit and the kit reliably detected the deletion (Supplementary Figure 1). In the present study, we planned – prior to sequencing (17 PCRs per patient) of EYA1 – to perform MLPA analysis (one ligation and one PCRs per patient), since deletions and duplications in EYA1 have been identified in up to 20% of EYA1 patients,3 and when applied in this order, the two methods provide the most time- and cost-efficient strategy for the molecular diagnosis of EYA1.
PCR and mutation analysis in EYA1 and SIX1
Primers were designed to amplify exons and surrounding intronic regions of EYA1 (GenBank nos. AJ000098 and NM_172060.1 (for exon 1)), SIX1 (GenBank no. NM_005982) and SIX5 (NM_175875.3), respectively (Supplementary Table 1). PCR-amplified samples were sequenced using the Big Dye Terminator kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. All 17 coding EYA1 exons were sequenced in family 1–6 and in addition EYA1 non-coding exons in family 4. Both coding exons of SIX1 in family 2 were sequenced. In order to study the segregation of the mutations with the disease in each family, we used either restriction enzyme digesting or sequencing. All detected mutations (Table 1) and polymorphisms (Supplementary Table 2) were investigated in at least 50 Danish control individuals, if the sequence variation was not reported before in the NCBI ENTREZ SNP database.
Prediction of consequences of splice site mutations
The effect of the identified splice site mutations on mRNA splicing was predicted by using the prediction server: Automated Splice Site Analysis (https://splice.cmh.edu/), which correlates the difference in information content (Ri) between the normal and the variant splice site sequence.23, 24 The minimal quantity of information required for splicing has been determined to be 2.4 bits.23
RT-PCR analysis
In order to investigate the effect of the splice site mutations IVS10−1G>A and IVS12+4A>G, respectively, we studied the expression of EYA1 in blood and skin. RT-PCR with primers amplifying exons 10–12 (forward: 5′-ccacttcagtttcccttgga-3′; reverse: 5′-ttacccgtctgtagcggaag-3′) and exons 8–13 (forward: 5′-gtcgaggttcagatgggaaa-3′; reverse: 5′-agtttgtccgggagtgaatg-3′), respectively, was performed on cDNA from blood and cultured fibroblasts. As control, PCR with internal primers (forward: 5′-ataactttggaacagatggc-3′; reverse: 5′-ctcttttacccgtctgtagc-3′) in exon 12 was performed. The PCR products were subsequently sequenced to verify the sequence amplified by the primers.
Results
Families
The identified mutations and the clinical data in the six BOR families are summarized in Table 1 and considerably more in detail in Supplementary Table 3. Below, the individual families are presented.
Family 1
The symptoms of the affected individuals in family 1 have previously been described.21 Briefly, the three affected individuals (II:6, III:3 and IV:4; Figure 1a) have hearing loss, branchial anomalies, preauricular pits and external ear deformity (representative audiograms are shown in Supplementary Figure 2). One individual (IV:1) who was otherwise healthy had external ear deformity. Moreover, one individual (IV:5) had congenital profound hearing impairment, using sign language (ie a very different hearing phenotype than the other affected individuals, see Supplementary Table 3), and no preauricular or branchial abnormalities and was therefore considered a phenocopy and unaffected with respect to BOR. Sequencing of GJB2 in this individual did not identify any mutations nor were the mutations del(GJB6-D13S1830) and del(GJB6-D13S1854) present in the patient. Renal ultrasound was performed as a follow-up in three individuals (III:3, IV:4 and IV:5), and turned out normal.
Family 1 is too small to obtain a statistically significant LOD score. Five members were included in linkage analysis for EYA1 and SIX1, and 12 individuals were included in linkage analysis to the BOR2 locus. The disease mutation seemed to be de novo in generation II, since individuals in generation I as well as the five siblings of the oldest patient (II:6) were reported to be healthy (Figure 1a). Furthermore, siblings of II:6 shared the disease haplotype with individual II:6 (data not shown), which supported the notion of a de novo mutation in II:6. The two-point LOD score did not reach statistical significance in favour of any of the loci.
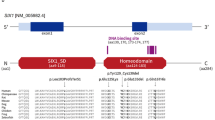
MLPA analysis of EYA1 was normal, but sequencing of EYA1 revealed a novel mutation, c.1773C>G, which results in the substitution of a tyrosine codon for a stop codon at position 591 (p.Y591X), two amino acids before the wild-type stop codon in EYA1 (Figure 2a). A restriction enzyme digest analysis confirmed the presence of the mutation in all three affected individuals (II:6, III:3 and IV:4), and the absence of the mutation in IV:5 and the other clinically unaffected individuals in the family (Figure 1a). The mutation was absent in 356 control chromosomes. Furthermore, the deleted Y591 and L592 are conserved in EYA1 from species such as mouse, chicken, frog and zebrafish (Figure 2b), and in the human EYA1 homologues EYA2 and EYA4. EYA3 has phenylalanine at position 591, which is nearly equivalent to a tyrosine residue. The evolutionary conservation of p.Y591 and p.L592, the co-segregation of the c.1773C>G mutation with the disease and the absence of the mutation in control individuals strongly indicate that this mutation underlies BOR in family 1.

(a) Sequence data for each of the mutations compared to a normal control. Each mutation is heterozygous in affected individuals. (b) Evolutionary conservation of EYA1 amino acids Y591 and L592. (c) Evolutionary conservation of SIX1 amino acid W122.
Family 2
Family 2 is reported for the first time as part of this study, and DNA was collected from eight individuals, four of whom were affected (V:5, VI:1, VI:2 and V:10; Figure 1b). Individual V:5 has progressive sensorineural hearing loss and unilateral preauricular pit and fistula. Two of her three children (VI:1 and VI:2) are affected and both have sensorineural hearing loss, but neither branchial nor preauricular anomalies. Representative audiograms are shown in Supplementary Figure 3. There were no subjective complaints indicating renal disease. In addition, psychiatric illness and mental retardation were reported in some family members (Supplementary Table 3). Inspection of the haplotypes did not exclude linkage to any of the three BOR loci. Neither MLPA analysis nor sequencing of EYA1 did reveal any abnormalities. By contrast, sequencing of SIX1 identified the sequence change c.364T>A in exon 1, leading to a substitution of tryptophan for arginine in position 122 of SIX1 (p.W122R) (Figure 2a). The mutation segregated with the disease in the family, and the mutation was absent in 140 control chromosomes as determined by sequencing. W122 is located in the Six domain of human SIX1 protein17 and is conserved in Six1 from species such as mouse, chicken and zebrafish and in the fly orthologue So (Figure 2c), as well as in all six human SIX homologues. The evolutionary conservation of W122, the co-segregation of the c.364T>A mutation with the disease and the absence of the mutation in control individuals strongly indicate that this mutation underlies BOR in family 2.
Family 3
Family 3 was initially reported in 1986 (family 3 in Gimsing and Dyrmose22) with classical BOR in five generations and affecting all organ systems (Table 1). Sixteen patients and seven unaffected at risk individuals as well as relevant spouses were included in linkage analysis. We obtained a significant two-point LOD score showing linkage to EYA1 and could exclude linkage to the BOR2 locus (meaning that the marker was not informative, but haplotype inspection excluded this locus) and to SIX1. MLPA analysis for EYA1 was negative, but sequencing of EYA1 revealed a splice site mutation in intron 10, IVS10−1G>A (1141−1G>A) (Figure 2a). The mutation involves the first nucleotide of the splice acceptor site in the intron and is therefore expected to result in aberrant splicing. Restriction-enzyme analysis showed that the mutation segregated with BOR in the family (Figure 1c). In one individual (V:11), the demonstration of the mutation confirmed a strong suspicion of BOR, since this 6-year-old boy had bilateral preauricular fistulae, but no detectable hearing impairment, so far. Renal ultrasound of the kidneys was unremarkable (Supplementary Table 3). The IVS10−1G>A mutation was not found in 100 control chromosomes. The nucleotide is conserved between species (http://genome.ucsc.edu/), suggesting a functional importance. Notably, the same nucleotide change has previously been reported in a BOR patient (patient MORL 2410),3 strongly implying that it is pathogenic.
In order to predict the effect of the IVS10−1G>A mutation on mRNA splicing, we used the prediction-server for splice mutations to calculate the Ri values for normal and variant nucleotide. The Ri value for the normal splice site in exon 11 declines from 6.3 → −16.1 bits, whereas the Ri for an activated new/cryptic splice site, one nucleotide upstream from the natural splice site, increases from −11.6 → 3.2 bits, which suggests that the new cryptic splice site is active, and the natural splice site becomes inactivated. Thus, the IVS10−1G>A mutation was predicted to move the splice site one nucleotide and introduce a frameshift at codon 380. Provided this mRNA is stable, it is expected to cause a protein with a disrupted phosphatase domain. To further examine the functional effect of this splice site mutation, we first established that EYA1 is expressed in fibroblasts, but not in blood, which agrees with previous findings.25, 26 We thereafter obtained a skin biopsy from an affected individual (IV:13) to obtain fibroblast cDNA from a person with the mutation. However, no variant band was detected by agarose gel electrophoresis and sequencing of RT-PCR products only detected mRNA with normal splicing. An explanation could be that the mRNA expressed from the ‘abnormal’ allele is unstable, whereby only the normal allele would be amplified by PCR.
Family 4
Family 4, briefly described in 1986 (family 1 in Gimsing and Dyrmose22), was updated as part of this study. There were no renal complaints, but otherwise typical features of BOR (Table 1). The family history indicated a de novo mutation in individual III:5 (Figure 1e). Her karyotype showed normal female chromosome constitution, 46, XX. Seventeen family members (five patients, eight at-risk individuals and four spouses) were included in the linkage analysis, and the two-point LOD scores indicated linkage to EYA1 for marker D8SS1795 (LODD8S1795=2.32), and seemed to exclude linkage to both the BOR2 locus and SIX1 (Table 2). The haplotype inspection raised strong suspicion of the possibility of a de novo EYA1 mutation in our proband's mother (Figure 1e; III:5). A re-calculation assuming such an event, excluding III:2, III:3, II:2 and II:3 gave a higher LOD score of 2.71 with marker D8S1795 at θ=0.0, which may be considered a significant LOD score once the disease locus is known.27 Sequencing of coding and non-coding exons in EYA1 only revealed polymorphisms (Supplementary Table 2) but no mutations. A new polymorphism (IVS9+107A>G) was introduced by a spouse and did not segregate with the disease. Furthermore, IVS9+107A>G was found in 16/56 normal controls, thereby being a frequent polymorphism in the Danish population. Forty-five base pairs in the 5′UTR of EYA1 (5′-AAACCAATAAGGTTAGGACAAGAGAATAGCTGTGGTTTGCGTTGC-3′) could not be sequenced. This stretch of DNA is located between two known polymorphisms (delT (rs35162278) and delAA (rs10635780, rs34976941)) found in the family. EYA1 MLPA analysis was normal, and a CGH study did not detect any submicroscopic imbalances (data not shown). Furthermore, SIX5 was sequenced in this family, but no disease mutation was identified. By these analyses we have so far been unable to identify the molecular cause of BOR in this family.
Family 5
Family 5 is a five-generation, large family with 13 patients and eight at-risk individuals available for linkage analysis. A previously description focussed on the occurrence of temporal bone abnormalities in 1991.6 All three BOR patients with EYA1 mutations (Figure 1f; individuals IV:10, IV:16 and V:12), who had a CT scan of the temporal bone reported in Ostri et al,6 shared the following abnormalities: hypoplastic middle ear cavity, severely malformed ossicular chain and hypoplastic cochleae, with only two coils and no evidence of enlarged endolymphatic duct.6 As part of the study we have extended the analysis of the family by including the youngest generation VI (Figure 1f). This family had end-stage renal disease, and otherwise typical BOR findings (Table 1). The linkage analysis with chromosome 8 markers showed linkage to EYA1 by two-point LOD score and multipoint LOD score and excluded linkage to both the BOR2 locus and SIX1. EYA1-MLPA analysis was normal, but sequencing of EYA1 revealed a splice mutation, IVS12+4A>G (c.1360+4A>G) (Figure 2a). Sequencing of DNA from the family members showed co-segregation of the mutation with the disease (Figure 1f). Sequencing of 226 control chromosomes for this splice site mutation did not detect the mutation. The nucleotide of the splice mutation is conserved in species such as mouse, dog, chicken and opossum (http://genome.ucsc.edu/), suggesting a functional importance. In contrast to the wild-type allele, the mutated allele was not detected by standard RT-PCR on cultivated fibroblasts from an affected individual (IV:14). By nested RT-PCR, however, we detected abnormally spliced EYA1 mRNA in the patient, in which exons 11 and 12 were deleted and a fragment of intron 10 was inserted instead. Taken together, these results indicate that the c.1360+4A>G splice site mutation results in abnormally spliced mRNA that is very unstable.
Family 6
Family 6 is a small three-generation family, published in 1986 (family 2 in Gimsing and Dyrmose22). Individual I:3 (maternal grandfather of our proband, III:3 in Figure 1d) had profound hearing loss, bilateral branchial sinuses and a unilateral preauricular pit. He had a unilateral hypoplastic kidney, cystic parenchyme and renal failure treated with dialysis from age 55. Furthermore, individual I:3 had a malformed cochlea and malformed ossicles and hypoplastic cavity of the middle ear. His daughter (II:2) had stationary sensorineural hearing loss, bilateral branchial sinuses, unilateral preauricular tag, bilaterally malformed ossicles and normal kidneys as determined from by intravenous urography. Her sister (II:3) died 3 h after delivery and had bilateral agenesis of the kidneys (Potter's syndrome). Individual II:2 has three children of whom one is affected (III:3). Individual III:3 had conductive hearing loss, unilateral branchial fistula, unilateral preauricular pit but no renal pathology as determined from a normal intravenous urography at age 7,22 and no subjective complaints at age 27 (Table 1). DNA was available from only four individuals and linkage analysis was therefore not feasible. EYA1 MLPA analysis was normal. By contrast, sequencing of EYA1 revealed a deletion, c.920delG, in exon 8 in EYA1 (Figure 2a). The deletion leads to a frameshift at codon 307 just before the phosphatase domain and a stop codon at amino acid 365. Restriction enzyme and sequencing analysis confirmed the co-segregation of the mutation with the disease in the family. The mutation was not present in 100 control chromosomes.
Discussion
In our study of six Danish BOR families, we identified the disease causing mutation in five families: four EYA1 mutations (c. 1773C>G, IVS10−1G>A, IVS12+4A>G and c.920delG) and one SIX1 mutation (c.364T>A).
The experience regarding SIX1 is very limited with only four different SIX1 mutations (in six unrelated families) known so far: Y129C,10, 17, 28 delE133,17, 29 R110W17 and now W122R, identified in our study. The W122R mutation, similar to the R110W and W122R mutation, is located in the SIX domain of the SIX1 protein, which is critical for the interaction with EYA1.17 The Y129C and the delE133 mutations are located in the homeodomain of the SIX1 protein, which is critical for DNA binding.17 Ruf et al17 showed the R110W mutation to reduce the affinity of the EYA1–SIX1 interaction, but not to affect the SIX1–DNA interaction, whereas the two homeobox mutations (Y129C and delE133) seemed to reduce both the SIX1–DNA and EYA1–SIX1 interactions. The W122R mutation in the SIX domain in our study is therefore expected to reduce the affinity for EYA1, but not for DNA.
Ruf et al17, 28 identified the Y129C missense mutation in the Anglo Saxon-Australian family underlying the report of the BOS3 locus, which has been used to describe BOR phenotype, but without renal anormalies (two patients in this family developed renal carcinomas, which was most likely coincidental). The same mutation was found in a large Japanese family with autosomal-dominant inheritance of mixed progressive hearing loss, unilateral EVA and unilateral ear pit, but no branchial and renal anomalies.10 Unfortunately, the authors only got permission to perform clinical and molecular studies of the proband.10 The missense mutation, R110W, was found in two unrelated German families, who had hearing loss, preauricular and branchial but no renal anomalies.17 Finally, the delE133 mutation was found in the same Swiss family, used for the identification of the non-syndromic hearing impairment, DFNA23 locus, who after re-investigation surprisingly turned out to have discrete renal anomalies.17, 29 In our family 2 with the W122R mutation, all affected members had hearing impairment and some had branchial and preauricular anomalies. Renal anomalies were not spontaneously reported in any affected individuals nor detected in three individuals analysed by ultrasound (Table 1).
Preliminary clinical experience based on the six-unrelated families with SIX1 mutation suggests mandatory presence of hearing impairment, a variable presence of EVA, low frequency (25%) of branchial arch malformations, lacrimal duct stenosis in about 10% and absence of renal pathology10, 17, 28, 29 (see also this study). One patient in our family 2 (individual IV:5; Figure 1b) underwent a CT scan before cochlear implant, but no malformations of the temporal bones were found. The frequent occurrence of temporal bone malformations in BOR patients in previous reports7, 8, 30 did not include verified mutations, but one study described a patient with a SIX1 mutation.10 The limited number of reported SIX1 mutations, so far, the low frequency of renal and branchial malformations in individuals who underwent examination for these abnormalities and lack of identified SIX1 mutations in eight Japanese BO patients may all indicate clinical differences in BO(R) patients with EYA1 and SIX1 mutations, respectively. However, more patients need to be studied in order to clarify this possibility.10 SIX1 mutations may be more prevalent in patients with hearing impairment and EVA, not caused by mutations in SLC26A4, than recognized so far. Therefore, we encourage more SLC26A4-negative patients with EVA and hearing impairment to be investigated for SIX1 mutations, in order to establish better genotype–phenotype correlations in clinically different patients.
Three of the four EYA1 mutations identified in this study (c.920delG, IVS10−1G>A and IVS12+4A>G) are predicted to lead to a protein lacking or with a disrupted phosphatase domain, (encoded by exons 9–16), and thereby in agreement with previous reports.3, 16, 31 Mutations in the phosphatase region are expected to lead to a non-functional protein, which is unable to bind to SIX1 and DACH. The mutation, c.920delG in exon 8, leads to a frameshift at codon 307, and thereby a protein lacking the phosphatase domain. Both splice mutations, IVS10−1G>A and IVS12+4A>G, were predicted to cause non-functional EYA1 protein with a disrupted phosphatase domain. The IVS10−1G>A splice mutation was previously reported by Chang et al3 in a BOR patient. This, together with the perfect co-segregation of the same mutation in our large family 3, provides strong evidence that the mutation is pathogenic. The only clinical difference was the absence of reported renal anomalies in the patient described by Chang et al,3 in contrast to our family 3 (Table 1), where 3 out of 7 analysed patients had renal pathology. Modifying factors, therefore, are suspected to play a role in determining the clinical implications of each mutation. Our family 5, previously reported with a main focus on ossicular chain abnormalities, hypoplastic cochleae and hypoplastic middle ear cavity, is now shown to be due to an EYA1 mutation (IVS12+4A>G).
The novel nonsense mutation in EYA1, c.1773C>G (p.Y591X) in exon 16 in family 1 is located just outside the phosphatase domain. Interestingly, Rickard et al31 identified a mutation (c1779A>C; X593Y(+5AA)) in the plus two codon relative to Y591 in a familial case (family 8 in Rickard et al31), and another nearby mutation (c.1748T>C; L583P) in a sporadic case (case 11 in Rickard et al31). The absence of our c.1773C>G mutation in 356 control chromosomes and the evolutionary conservation of this region in several species and conservation in EYA1–4 also point to a functional role of Y591 and that the Y591X mutation is pathogenic.
The absence of EYA1 mutation in family 4 is a puzzle considering the linkage results in favour of EYA1 with a two-point LOD score of 2.32 increasing to 2.71 if the hypothesis of a de novo mutation in the probands mother (Figure 1e, III:5) is taken into account. Some investigators have previously suggested that mutations in NDUFB9, in the same region (8q13), could cause a BOR phenotype;32 however, they did not find any NDUFB9 mutations in nine BOR families. Another explanation might be a mutation in EYA1 regulatory elements (conserved-nongenic-elements) close to EYA1, leading to an inactivation of EYA1 transcription. There does not seem to be any CpG island near EYA1; hence methylation differences are not likely. Mutations in non-coding exons in EYA1, which could be involved in gene regulation, might be another explanation, but the normal sequence results for the non-coding exons did not support this possibility in family 4.
Our report adds to the accumulating genotype–phenotype correlations in BOR. The figures in Table 1 are minimum estimates regarding subtle and subclinical findings, because we only collected already available information about renal and temporal bone abnormalities in these families. The importance of clinical evaluation and molecular testing in BOR has been shown by a number of authors testing both isolated and familial cases.31 The clinical presentation of SIX1-related disease may even mimick non-syndromic hearing impairment, as illustrated in the DFNA23 family, who turned out to have SIX1 mutation and mild BOR pathology.17, 29 The detection rate of mutations in EYA1 and SIX1 in 5 out of 6 probands in this study are encouraging for sequencing EYA1 and SIX1 in clinically suspected patients, irrespective of a positive family history.31, 33 The diagnosis of Potter's syndrome in newborn, as illustrated by individual II:3 in family 6 (Figure 1d), should also elicit a suspicion of BOR and sequencing of EYA1 and SIX1.
Recently, we reported another Danish family with BOR syndrome caused by a novel splice site mutation (IVS9+1G>C) in EYA1.25 In total, we identified an EYA1 or a SIX1 mutation in 6 out of 7 (86%) Danish BOR families by direct sequencing (see this study and Henriksen et al25). Our detection rate of mutations is comparable to the 71% reported in a series of Japanese patients (EYA1 mutation in 5 out of 7 patients, 2 familial and 5 sporadic, but no SIX1 mutations), also applying direct sequencing of both BOR genes33 and the 61% in 18 patients (11 familial and 7 sporadic) in an English study using SSCP and sequencing of EYA1 only.31 Several previous studies reported a much lower detection rate of EYA1 mutations of 25–40% in BOR patients with the highest success rate in familial cases.3, 15, 34, 35 The absence of EYA1 abnormalities detectable by MLPA, which was applied for the first time in the present study, does not support the previous suggestion of a 20% frequency of complex rearrangements of EYA1 as causative in BOR.15 More reliable figures for the proportion of patients having deletions or duplications, detectable by MLPA, and the proportion having BOR due to SIX1 mutations can only be reached by studying more patients.
In conclusion, we identified disease causing EYA1 and SIX1 mutations in 5 out of 6 Danish families comprising 66 patients, and strongly encourage to look for mutations in clinically suspicious cases. A novel SIX1 mutation adds to the few cases reported so far. Our data provide substantial molecular and clinical information for establishing a better genotype–phenotype understanding of BOR. Larger studies including patients with hearing impairment and EVA, but no branchial or renal malformations, will clarify if the preliminary experience of different clinical picture of SIX1- and EYA1-related disease is consistent.
References
Melnick M, Bixler D, Silk K, Yune H, Nance WE : Autosomal dominant branchiootorenal dysplasia. Birth Defects Orig Artic Ser 1975; 11: 121–128.
Chen A, Francis M, Ni L et al: Phenotypic manifestations of branchio–oto–renal syndrome. Am J Med Genet 1995; 58: 365–370.
Chang EH, Menezes M, Meyer NC et al: Branchio–oto–renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat 2004; 23: 582–589.
Fraser FC, Sproule JR, Halal F : Frequency of the branchio–oto–renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet 1980; 7: 341–349.
Kemperman MH, Koch SMP, Kumar S, Huygen PLM, Joostein FBM, Cremers CWRJ : Evidence of progression and fluctuation of hearing impairment in branchio–oto–renal syndrome. Int J Audiol 2004; 43: 523–532.
Ostri B, Johnsen T, Bergmann I : Temporal bone findings in a family with branchio–oto–renal syndrome (BOR). Clin Otolaryngol 1991; 16: 163–167.
Propst EJ, Blaser S, Gordon KA, Harrison RV, Papsin BC : Temporal bone findings on computed tomography imaging in branchio–oto–renal syndrome. Laryngoscope 2005; 115: 1855–1862.
Ceruti S, Stinckens C, Cremers CW, Casselman JW : Temporal bone anomalies in the branchio–oto–renal syndrome: detailed computed tomographic and magnetic resonance imaging findings. Otol Neurotol 2002; 23: 200–207.
Kemperman MH, Stinckens C, Kumar C, Joostein FBM, Huygen PLM, Cremers CWRJ : The branchio–oto–renal syndrome. In: Cremers CWRJ, Smith RJH (eds),: Genetic Hearing Impairment. Basel: Karger, Adv Otorhinol. 2002; 61: 192–200.
Ito T, Noguchi Y, Yashima T, Kitamura K : SIX1 mutation associated with enlargement of the vestibular aqueduct in a patient with branchio–oto syndrome. Laryngoscope 2006; 116: 796–799.
Kochhar A, Fischer SM, Kimberling WJ, Smith RJ : Branchio–oto–renal syndrome. Am J Med Genet A 2007; 143: 1671–1678.
Hereditary Hearing loss Homepage, http://webhost.ua.ac.be/hhh.
Kumar S, Deffenbacher K, Marres HAM, Cremers CWRJ, Kimberling WJ : Genomewide search and genetic localization of a second gene associated with autosomal dominant branchio–oto–renal syndrome: clinical and genetic implications. Am J Hum Genet 2000; 66: 1715–1720.
Hoskins BE, Cramer CH, Silvius D et al: Transcription factor SIX5 is mutated in patients with branchio–oto–renal syndrome. Am J Hum Genet 2007; 80: 800–804.
Vervoort VS, Smith RJ, O’Brien J et al: Genomic rearrangements of EYA1 account for a large fraction of families with BOR syndrome. Eur J Hum Genet 2002; 10: 757–766.
Web source (IOWA database) listing identified mutations in EYA1, http://www.medicine.uiowa.edu/pendredandbor/eya1Mutations.htm.
Ruf RG, Xu P-X, Silvius D et al: SIX1 mutations cause branchio–oto–renal syndrome by disruption of EYA1–SIX–DNA-complexes. Proc Natl Acad Sci USA 2004; 101: 8090–8095.
Rebay I, Silver SJ, Tootle TL : New vision from Eyes absent: transcription factors as enzymes. Trends Genet 2005; 21: 163–171.
Li X, Oghi KA, Zhang J et al: Eya protein phosphatase activity regulates Six1–Dach–Eya transcriptional effects in mammalian organogenesis. Nature 2003; 426: 247–254.
Ohto H, Kamada S, Tago K et al: Cooperation of Six and Eya in activation of their target genes through nuclear translocation of Eya. Mol Cell Biol 1999; 19: 6815–6824.
Gimsing S : The BOR syndrome as a possible neurocristopathy. Ear, Nose Throat J 1987; 66: 154–158.
Gimsing S, Dyrmose J : Branchio–oto–renal dysplasia in three families. Ann Otol Rhinol Laryngol 1986; 95: 421–426.
Rogan PK, Faux BM, Schneider TD : Information analysis of human splice site mutations. Hum Mutat 1998; 12: 153–171.
Nalla VK, Rogan PK : Automated splicing mutation analysis by information theory. Hum Mutat 2005; 25: 334–342.
Henriksen AM, Tümer Z, Tommerup N, Tranebjærg L, Larsen LA : Identification of a novel EYA1 splice-site mutation in a Danish branchio–oto–renal syndrome family. Genet Test 2004; 8: 404–406.
Expression-database, NCBI, Unigene, EST profile viewer, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene.
Ott J : Analysis of Human Genetic Linkage. Baltimore and London: The John Hopkins University Press, 1999, pp 260–261.
Ruf RG, Berkman J, Wolf MTF et al: A gene locus for branchio–otic syndrome maps to chromosome 14q21.3-q24.3. J Med Genet 2003; 40: 515–519.
Salam AA, Häfner FM, Linder TE, Spillmann T, Schinzel AA, Leal SM : A novel locus (DFNA23) for prelingual autosomal dominant nonsyndromic hearing loss maps to 14q21-q22 in a Swiss German kindred. Am J Hum Genet 2000; 66: 1984–1988.
Stinckens C, Standaert L, Casselman JW et al: The presence of a widened vestibular aqueduct and progressive sensorineural hearing loss in the branchio–oto–renal syndrome. A family study. Int J Pediatr Otorhinol 2001; 59: 163–172.
Rickard S, Boxer M, Trompeter R, Bitner-Glindzicz M : Importance of clinical evaluation and molecular testing in the branchio–oto–renal (BOR) syndrome and overlapping phenotypes. J Med Genet 2000; 37: 623–627.
Lin X, Wells DE, Kimberling WJ, Kumar S : Human NDUFB9 gene: Genomic organization and a possible candidate gene associated with deafness disorder mapped to chromosome 8q13. Hum Hered 1999; 49: 75–80.
Okada M, Fujimaru R, Morimoto N et al: EYA1 and SIX1 gene mutations in Japanese patients with branchio–oto–renal (BOR) syndrome and related conditions. Pediatr Nephrol 2006; 21: 475–481.
Abdelhak S, Kalatzis V, Heilig R et al: Clustering of mutations responsible for branchio–oto–renal (BOR) syndrome in the eyes absent homologous (eysHR) of EYA1. Hum Mol Genet 1997; 6: 2247–2255.
Migliosi V, Flex E, Guida V et al: Identification of five novel BOR mutations in human EYA1 gene associated with branchio–oto–renal syndrome by a DHPLC-based assay. Clin Genet 2004; 66: 478–480.
Acknowledgements
We thank the families who participated in this study. In addition, we acknowledge the skilful guidance and technical assistance of Annemette Friis Mikkelsen, Lillian Egebjerg Rasmussen and Karen Friis Henriksen, and valuable technical help from Elvira Chapka and Marianne Lodahl at Department of Cellular and Molecular Medicine, ICMM, The Panum Institute. We also thank laboratory technician Anne-Lise Palle at the University Hospital, Rigshospitalet, Copenhagen, for performing the MLPA analysis, and Stephanie Fischer and Richard Smith, Department of Otolaryngology Head and Neck Surgery, University of Iowa Medical School, Iowa City, USA, for providing control samples for the MLPA analysis. The project was hosted by Wilhelm Johannsen Centre for Functional Genome Research, which is funded by the Danish National Research Foundation. The study was supported by the Oticon Foundation and Widex AS (NDR, LT and KMS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Sanggaard, K., Rendtorff, N., Kjaer, K. et al. Branchio–oto–renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet 15, 1121–1131 (2007). https://doi.org/10.1038/sj.ejhg.5201900
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201900
Keywords
This article is cited by
-
Auricular fistula: a review of its clinical manifestations, genetics, and treatments
Journal of Molecular Medicine (2023)
-
Phenotype–genotype correlation in patients with typical and atypical branchio-oto-renal syndrome
Scientific Reports (2022)
-
A novel mutation in EYA1 in a Chinese family with Branchio-oto-renal syndrome
BMC Medical Genetics (2018)
-
Recurrent 8q13.2-13.3 microdeletions associated with Branchio-oto-renal syndrome are mediated by human endogenous retroviral (HERV) sequence blocks
BMC Medical Genetics (2014)
-
Identification of a novel nonsynonymous mutation of EYA1 disrupting splice site in a Korean patient with BOR syndrome
Molecular Biology Reports (2014)
