
Abstract
The human ZIC3 gene has been mapped to Xq26.2, the visceral heterotaxy locus HTX1, and has been shown to be mutated in X-linked situs ambiguus and/or complex heart defects. We report on a female fetus with situs ambiguus, asplenia and corrected transposition of the great arteries, displaying a (X;21) translocation. The balanced state of the t(X;21)(q26;p13) was verified by FISH on metaphase chromosomes of the fetus using DOP-PCR products of the microdissected der(21) and Xq-subtelomere specific sequences, and by PRINS with β-satellite specific sequences. Examination with polymorphic markers flanking ZIC3 on DOP-PCR products of the microdissected der(21) chromosome evidenced that the complete copy of the ZIC3 gene was translocated to chromosome 21. Mutations in the fetal and parental ZIC3 genes were excluded by sequencing. Paternal origin of the der(X) and der(21) chromosomes was confirmed by use of polymorphic microsatellite markers from chromosome 21 and from the chromosomal region Xq26, respectively. X chromosome inactivation analysis using a PCR of a polymorphic (CAG)n repeat in the first exon of the androgen receptor gene showed a completely skewed X inactivation pattern with the paternal X as the active X chromosome, thus excluding functional disomy of distal Xq. A positional effect caused by the balanced (X;21) translocation may be responsible for functional nullisomy of ZIC3 and thus explain the situs and cardiac abnormalities in the fetus.
Similar content being viewed by others


Introduction
Laterality is determined by a number of autosomal and X-chromosomal genes. Mutations may cause situs inversus totalis or partialis (=situs ambiguus). While situs inversus (totalis) stands for complete reversal of the normal left–right asymmetry of viscera, generally not coinciding with cardiac defects, situs ambiguus designates incomplete reversal and randomization of organ position with either preferential right sidedness (asplenia, bilaterally three lobed lungs, complex cardiac defects) or preferential left sidedness (polysplenia, bilaterally bilobed lungs, less severe cardiac defects). One of the genes for situs abnormalities in man, the heterotaxy locus HTX1, was mapped to human chromosome Xq26.2.1,2 In this region, the human ZIC3 gene was localized by positional cloning. Loss of function mutations in ZIC3 was found to be associated with familial or sporadic situs ambiguus and/or complex heart defects.3,4,5
ZIC3 encodes a five C2H2-containing zinc-finger transcription factor and shows homology to the Drosophila pair-rule gene odd paired (opa).6,7 In Xenopus and mouse, Zic3 plays a role in neural and neural crest development and participates in the left signaling pathway.8,9 Deletions of the zinc-finger or N-terminal domains of Zic3 as well as right-sided Zic3 overexpression in Xenopus result in L–R axis disturbance and alter heart and gut looping, thus causing situs abnormalities.10 Deletion of the murine Zic3 is known as mouse mutant ‘bent tail’ (Bn), which showed a short kinked tail besides situs abnormalities and, in about 10% of the nonviable males, exencephaly and other defects.11,12
We present the first case of a balanced (X;21) translocation involving a nonmutated but potentially inactivated ZIC3 gene region, leading to situs ambiguus with asplenia, corrected transposition of great arteries, anal anomalies and sacral hypoplasia – all malformations seen in Zic3 mutations of Xenopus, mouse and man, respectively.
Case report
A 37 year-old gravida 2 para 0 was referred for amniocentesis because of advanced maternal age. The paternal age was 38 years. A preceding pregnancy had been terminated for personal reasons. The family history was unremarkable and with no evidence for consanguinity. The fetal karyotype showed additional Xq-material on the short arm of chromosome 21, which was primarily interpreted as an unbalanced de novo translocation. Ultrasound examination revealed a complex cardiac malformation and situs inversus of the fetus. The parents decided for termination of pregnancy, which was carried out at 18+1 weeks of gestation.
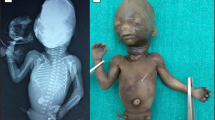
The female fetus of 230 g weight and 15.5 cm/22.5 cm crown-rump/crown-heel length was severely macerated. It displayed mild dysmorphic features with a relatively large, slightly turricephalic skull, high front, short flat and narrow nose with broad prominent root, prominent philtrum, macrostomia with broad lips, distinct micro-retrognathia and small, poorly modulated ears (Figure 1a). There were bilateral single palmar creases and mild caudal hypoplasia with slightly shortened legs, hypoplastic genitalia and ectopic anus (Figure 1b). X-ray revealed sacral hypoplasia (Figure 1c). At autopsy, a situs ambiguus with asplenia, bilaterally four-lobed lungs, malrotation of stomach and intestine, midline position of heart, liver, mobile cecum and descending colon were found. The heart showed rotation to the left and a complex malformation with a persistent common atrio-ventricular (AV) canal, a corrected transposition of the great arteries with a pseudotruncus communis aortalis arising from a right-sided, functionally left ventricle. A severely stenotic pulmonic artery of 1 mm diameter, arising from the left-sided right ventricle, inflow of the caval veins into the left-sided atrium and inflow of the pulmonic veins into both atria and a right descending aorta was also found. There were no anomalies of the urogenital organs and no gross malformations of the brain.

(a–c) Severely macerated female fetus of 18+1 gestational weeks showing (a) mildly dysmorphic features with high front, short flat nose, broad nasal root, prominent philtrum, macrostomia, thick lips, microretrognathia, small ears and (b) genital hypoplasia and an ectopic, ventrally placed anus. (c) X-ray revealed 11 ribs and sacral hypoplasia, with only two ossified vertebral bodies.
Cytogenetic and FISH analyses
Conventional cytogenetic analysis (GTG banding) of amniotic fluid cells at 18 gestational weeks revealed a derivative chromosome 21 (der(21)) containing additional genetic material on its short arm, which was identified to originate from the X-chromosome by M-FISH. This finding was initially interpreted as an unbalanced (X;21) translocation causing functional disomy of Xq26 → qter. The derivative chromosome 21 was confirmed in chorionic villi after induced abortion. However, one of the X chromosomes appeared slightly shortened. Therefore, a balanced state of the translocation was suspected. Cytogenetic analyses of peripheral blood lymphocytes of the healthy parents were normal. Chromosomal mosaicism was excluded in the father's lymphocytes by analysis of 150 metaphases.
In order to determine the chromosome aberration of the fetus more precisely, chromosome microdissection was performed.13 In three approaches five derivative chromosomes 21 were microdissected and amplified by DOP-PCR using three different Taq polymerases (AmpliTaq DNA Polymerase, AmpliTaq Gold DNA Polymerase, PE Applied Biosystems, Inc., Lincoln, CA, USA; DOP-PCR Master Kit, Roche Mannheim, Germany). The specific chromosome libraries were subsequently used for FISH to metaphases of the fetus as well as for microsatellite analysis. The FISH results showed signals covering the whole derivative chromosome 21 and the long arm of the normal chromosome 21 (Figure 2a). A distinct signal was detected at the distal long arm of the slightly larger X chromosome comprising the region Xq26-qter. There was no hybridization signal on the slightly shorter X-chromosome. Thus, the suspected balanced state of the t(X;21) was confirmed.

(a–d) Molecular-cytogenetic (FISH) analyses of the fetal rearrangement, confirming a reciprocal translocation between Xq and 21p. (a) Chromosome painting with the microdissected aberrant chromosome 21 as a probe to fetal metaphases (forward painting), revealing a distinct hybridization signal in distal Xq on the normal X chromosome, whereas the derivative X chromosome shows no signals. The entire der(21) as well as the long arm of the normal chromosome 21 are painted. (b) PRINS with the β-satellite sequences yielded detectable sequences in the derivative X chromosome. (c) Silver staining of the NOR regions indicating a very distal breakpoint in der(21p). (d) Dual-color FISH with a subtelomeric probe of Xq (red signals) and a chromosome X centromere specific probe (green signals) showing translocation of Xq to 21p.
Primed in situ labeling (PRINS) was performed essentially as described by Koch et al.14 Oligonucleotides representing satellite III and β-satellite sequences were used as primers (Vector Lab. Burlingame, CA, USA). PRINS with β-satellite specific sequences showed a distinct signal on the distal long arm of the derivative X chromosome (Figure 2b), while PRINS with the satellite III probe revealed no detectable signal in Xq (data not shown). There was no signal on the X chromosome after NOR staining (Figure 2c). Due to the relative locations of the different classes of tandemly repeated DNAs on the short arm of the acrocentric chromosomes (α-satellite, β-satellite, satellite III, rDNA, β-satellite, cytological satellite15), the breakpoint on the short arm of chromosome 21 must be distal of rDNA, most probably in chromosome band 21p13.
FISH studies using a subtelomeric probe specific for the locus DXYS129 (Vysis Inc., Downers Grove, IL, USA) and an X chromosome specific centromere probe (DXZ1) were applied according to the manufacturer's protocols. Xq-subtelomere specific sequences were deleted at the telomere of the long arm of the abnormal X chromosome and were visualized at the short arm of the der(21) (Figure 2d).
According to these data, a balanced reciprocal translocation between chromosomes X and 21 was confirmed and the fetal karyotype was determined as 46,X,t(X;21)(q26;p13).
DNA analyses
Genomic DNA from frozen fetal tissue and chorionic villus cell samples and from leucocytes from the parents' peripheral blood was isolated by the standard salting-out procedure.
Mutation analysis of the ZIC3 gene was performed on fetal and parental genomic DNA. For gene amplification, primer sets and PCR conditions were chosen as described by Gebbia et al.3 PCR products were purified by Gel Extraction Kit (Qiagen GmbH, Hilden, Germany). Approximately 50 ng of PCR products were bidirectionally sequenced using a sequencing kit (BigDye terminator; PE Applied Biosystems) and were analyzed on an automated DNA sequencer (ABI 310; PE Applied Biosystems) according to the manufacturer's protocols. Sequence alignments were performed with the ‘Sequencher’ software package (Gene Codes, Ann Arbor, MI, USA). No aberrant sequence was detected, neither in the fetal nor in the parental DNA, thus excluding missense, nonsense or frameshift mutations in the coding region of ZIC3.
PCR reactions on DOP-PCR products obtained from the microdissected der(21) of the fetus were performed using primer pairs of ZIC3 flanking polymorphic markers DXS8050 and DXS1041. Both PCR reactions showed specific products confirming the localization of ZIC3 on the derivative chromosome 21 (Figure 3, Table 1).

(a) Partial karyogram showing G-banded homologues of chromosomes X and 21 with ideograms representing the t(X;21)(q26;p13) based on the FISH results. The schematic representation of the ZIC3 gene region is shown on the right side, with microsatellite markers DXS8050 and DXS1041 flanking ZIC3. (b) Verification of the localization of the ZIC3 gene region on the der(21) by PCR with genomic DNA from the fetus and the parents, as well as DOP-PCR amplified DNA from the microdissected der(21) used as template. Lanes 1–3 represent three different DOP-PCR amplicons of the der(21) chromosome.
PCR reactions on DOP-PCR products using polymorphic markers from the chromosomal region Xq26 (DXS1041, DXS8050, DXS8091, DXS1193, DXS8011, Research Genetics Inc., Huntsville, AL, USA) and 21q (D21S1432, D21S156, D21S1435, D21S1270) were aimed to determine the parental origin of the derivative chromosome 21. While most of the markers were uninformative in this family, DXS1193 and D21S1270 were informative, showing paternal origin of the der(21) and the translocated Xq segment (Table 1).
X inactivation analysis
X inactivation pattern was determined by PCR analysis of a polymorphic (CAG)n repeat in the first exon of the androgen receptor (AR) gene.16After digestion of DNA with the methylation-sensitive enzyme HpaII, a PCR product was obtained from the inactive X chromosome only. The PCR products were separated on an ABI 377 automated sequencer, and analyzed by GeneScan software (PE Applied Biosystems). The X inactivation phenotype was recorded as the ratio between the two PCR products with the smallest allele given first.
The X inactivation pattern is shown in Figure 4. The fetus had a completely skewed X inactivation pattern in fetal tissues (100:0) with the paternal X as the preferentially active X chromosome, whereas the X inactivation in chorionic villi was random. The mother had a 20:80 pattern, with the AR allele that was inherited by the fetus on the predominating inactive X chromosome.17

X inactivation analysis. HpaII−: undigested DNA, HpaII+: digested DNA. The ratios indicate the PCR products from the inactive X chromosome, with the smallest allele first. Note completely skewed X inactivation in fetal tissue, and lack of PCR product from the father.
Discussion
We present a female fetus with a 46,X,t(X;21)(q26;p13) karyotype and abnormal phenotype. The fetus has a paternally derived (X;21) translocation where the complete ZIC3 locus is preserved on the distal short arm of chromosome 21 in vicinity to the NOR region. The paternal der(X) chromosome is 100% active in the fetus and the maternal normal X is completely inactive. The fetus has no mutation in ZIC3, but has all clinical signs compatible with a nullisomy of the whole ZIC3 locus.
X-linked laterality sequence has been well documented in the past.18,19,20 A heterotaxy locus HTX1 representing the human ZIC3 gene has been cloned and mapped to Xq26.1,2 Regardless of the specific ZIC3 mutations identified to date, all but two affected males are situs inversus with variable combinations of complex heart disease, anal anomalies, sacral agenesis and spina bifida. Heterozygous females had milder malformations including situs ambiguus and anal anomalies.3,21 Recently, ZIC3 mutations have also been identified in patients with isolated congenital heart disease.21 The cardiac malformations comprised a combination of anomalies of the venous inflow and arterial outflow tract, such as single atrium, single ventricle, persistent atrioventricular canal (AVC), VSD or ASD (corrected), transposition of the great arteries (TGA) or right descending aorta, pulmonic stenosis or atresia (PS) and often dextroposition of the heart and right descending aorta. Our case displaying right-sided situs ambiguus with asplenia, bilaterally four-lobed lungs, malrotation of the intestine and a characteristic heart defect (AVC+TGA+PS) and also anal ectopia and sacral hypoplasia fits well into the malformation spectrum of the above male cases, with the exception that it lacked a mutation in ZIC3 and that it concerned a female with a balanced 46,X,t(X;21)(q26;p13) chromosome translocation.
Associated sacral and anal anomalies in our case may be considered as a phenotypical equivalent of the short-kinked tail caused by deletion of Zic3 in the mouse mutation ‘bent tail’ (Bn).22 This may differentiate X-linked situs abnormalities from autosomal dominant visceral heterotaxia types HTX2, characterized by loss of function mutations in the EGF-CFC gene CFC1,23 and HTX3 with a gene locus mapped to 6q21.24,25 Recently, mutations in the cell adhesion molecule CRELD1 and NKX2.5 also have been associated with heterotaxy.26,27 However, only single patients with laterality defects were observed and the phenotypic consequences of gene defects in these loci therefore require further characterization.
The finding of an affected female may reflect skewed X inactivation within the fetus. The fetus had a completely skewed X inactivation, with the abnormal paternal X chromosome being the 100% active and the normal maternal X chromosome the inactive one. In balanced female carriers, the normal X chromosome is usually inactivated, leaving the derivative chromosome X in an active state.28,29,30 This is attributed to selective growth of cells that have inactivated the normal X, thus having a balanced dosage of expressed genes. Approximately 77% of females with balanced X/autosome translocations exhibit complete nonrandom inactivation of the normal X chromosome. These cases have the breakpoints distributed all along the X chromosome, but there is no comparable case listed with breakpoints affecting distal Xq and short arms of an acrocentric chromosome,31 making our case a first example. In about 23% of X/autosome translocation carriers, the derivative X chromosome is inactivated. Interestingly, in these cases, the breakpoints tend to be located in the telomeric regions of Xp and Xq, demonstrating that skewed X inactivation might depend on the localization of the breakpoint.
Remarkably, a random X inactivation pattern was found in chorionic villi. This finding supports the hypothesis of strong selection against cells with an inactivation of the translocated X chromosome in most of the embryonic/fetal tissues and a weaker selection force taking place during placental development.32
Overall, these findings suggest that loss of function of ZIC3 may underlie the pathogenesis in the fetus. One of the factors determining incorrect ZIC3 gene expression might be disruption from upstream X-chromosomal regulating (eg enhancing) genes, leading to reduction or absence of gene transcription. This has been shown to play a role in a number of human genetic diseases with breakpoints at some distance from the causative gene.33 In several model organisms, the apposition of expressed genes next to heterochromatin is known to cause position effects that result in silencing. In particular, it has been suggested that the promoter region of an X-linked gene could be less accessible to enhancer elements when embedded in heterochromatin.34 Juxtaposition of the euchromatic ZIC3 gene to a region of heterochromatin on the short arm of chromosome 21 might be another cause for inhibition of gene expression. Spreading of heterochromatization into the juxtaposed euchromatin may give rise to position effect variegation, as has been shown in chromosomal rearrangements of Drosophila and mammalian systems.35,36,37 Otherwise, movement of a euchromatic region to a transcription-incompetent area of the nucleus38,39,40 may reduce the expression of genes. The fetus shows no additional anomalies, although there must be X-chromosomal genes other than ZIC3 involved in the translocation. We may presume that heterochromatization affects chromosome regions directly adjacent to the breakpoint in chromosome 21p13, such as the ZIC3 gene, or the whole translocated distal Xq region. But, in the latter case, heterochromatization of genes like color blindness or the FRAX gene would not have been diagnosed in the aborted fetus.
The assumption of functional nullisomy of the translocated ZIC3 through position effect silencing in our case is supported by an observation of a nearly identical but presumably unbalanced 46,XY,der(21)t(X;21)(q26;p11.2) translocation in a boy, leading to functional disomy of the translocated Xq segment.41 The boy presented with pre- and postnatal growth retardation, developmental delay, hypotonia, microcephaly, agenesis of the corpus callosum, dysmorphic facial features, cryptorchism and left multidysplastic kidney, but not with situs ambiguus, cardiac malformation and sacral or anal anomalies. Since the latter anomalies have been ascribed to loss-of-function mutations of ZIC3 in man and in addition to gain-of-function mutations in Xenopus,8 this might indicate that in this case ZIC3 in the translocated Xq segment is inactivated by the same mechanisms, as discussed above, thus excluding it from the functional disomy of the remainder of the translocated Xq segment.
References
Casey B, Devoto M, Jones KL, Ballabio A : Mapping a gene for familial situs abnormalities to human chromosome Xq24–q27.1. Nat Genet 1993; 5: 403–407.
Ferrero GB, Gebbia M, Pilia G et al: A submicroscopic deletion in Xq26 associated with familial situs ambiguus. Am J Hum Genet 1997; 61: 395–401.
Gebbia M, Ferrero GB, Pilia G et al: X-linked situs abnormalities result from mutations in ZIC3. Nat Genet 1997; 17: 305–308.
Mégarbané A, Salem N, Stephan E et al: X-linked transposition among males with a nonsense mutation in ZIC3. Eur J Hum Genet 2000; 8: 704–708.
Ware S, Peng J, Zhu L et al: Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet 2004; 74: 93–105.
Benedyk MJ, Mullen JR, DiNardo S : Odd-paired: a zinc finger pair-rule protein required for the timely activation of engrailed and wingless in Drosophila embryos. Genes Dev 1994; 8: 105–117.
Aruga J, Nagai T, Tokuyama T et al: The mouse Zic gene family: homologues of the Drosophila pair-rule gene odd-paired. J Biol Chem 1996; 271: 1043–1047.
Kitaguchi T, Nagai T, Nakata K, Aruga J, Mikoshiba K : Zic3 is involved in the left–right specification of the Xenopus embryo. Development 2000; 127: 4787–4795.
Nagai T, Aruga J, Takada S et al: The expression of the mouse Zic1, Zic2 and Zic3 gene suggests an essential role for Zic genes in body pattern formation. Dev Biol 1997; 182: 299–313.
Nakata K, Nagai T, Aruga J, Mikoshiba K : Xenopus ZIC3, a primary regulator both in neural and neural crest development. Proc Natl Acad Sci USA 1997; 94: 11980–11985.
Carrel T, Purandare SM, Harrision W et al: The X-linked mouse mutation Bent tail is associated with a deletion of the Zic3 locus. Hum Mol Genet 2000; 9: 1937–1942.
Klootwijk R, Franke B, van der Zee CEEM et al: A deletion encompassing Zic3 in Bent tail, a mouse model for X-linked neural tube defects. Hum Mol Genet 2000; 9: 1615–1622.
Friedrich U, Houman M, Holm-Hansen B, Kaltoft K : Microdissection and reverse painiting in a melanoma cell line: a detailed description of structurally abnormal chromosomes. Cancer Genet Cytogenet 2001; 125: 5–9.
Koch J, Hindkjaer J, Kolvraa S, Bolund L : Construction of panel of chromosome-specific oligonucleotide probes (PRINS-primer) useful for the identification of individual human chromosomes in situ. Cytogenet Cell Genet 1995; 71: 142–147.
Gravholt CH, Friedrich U, Caprani M, Jørgensen AL : Breakpoints in Robertsonian translocations are localized to satellite III DNA by fluorescense in situ hybridization. Genomics 1992; 14: 924–930.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW : Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–1239.
Ørstavik KH, Ørstavik RE, Naumova AD et al: X chromosome inactivation in carriers of Barth syndrome. Am J Hum Genet 1998; 63: 1457–1463.
Soltan HC, Li MD : Hereditary dextrocardia associated with other congenital heart defects: report of a pedigree. Clin Genet 1974; 5: 51–58.
Mathias RS, Lacro RV, Jones KL : X-linked laterality sequence: situs inversus, complex cardiac defects, splenic defects. Am J Med Genet 1987; 28: 111–116.
Mikkilä SP, Karikowski TT, Simola KOJ : X-linked laterality sequence in a family with carrier manifestations. Am J Med Genet 1994; 49: 435–438.
Ware SM, Peng J, Zhu L et al: Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet 2004; 74: 93–105.
Herman GE, El-Hodiri HM : The role of Zic3 in vertebrate development. Cytogenet Genome Res 2002; 99: 229–235.
Bamford RN, Roessler E, Burdine RD et al: Loss-of-function mutations in the EGF–CFC gene CFC1 are associated with human left–right laterality defects. Nat Genet 2000; 18: 301–302.
Kato R, Yamada Y, Niikawa N : De novo balanced translocation (6;8)(q21;q21.3) in a patient with heterotaxia. Am J Med Genet 1996; 66: 184–186.
Peeters H, Debeer P, Groenen P et al: Recurrent involvement of chromosomal region 6q21 in heterotaxy. Am J Med Genet 2001; 103: 44–47.
Robinson SW, Morris CD, Goldmuntz E et al: Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet 2003; 72: 1047–1052.
Watanabe Y, Benson DW, Yano S et al: Two novel frameshift mutations in NKX2.5 result in novel features including visceral inversus and sinus venosus type ASD. J Med Genet 2002; 39: 807–811.
Brown CJ, Robinson W : The causes and consequences of random and nonrandom X chromosome inactivation in humans. Clin Genet 2000; 58: 353–363.
Zabel BU, Baumann WA, Pirntke W, Gerhard-Ratschow K : X-inactivation pattern in three cases of X/autosome translocation. Am J Med Genet 1978; 9: 316–320.
Belmont JW : Genetic control of X inactivation and processes leading to X-inactivation skewing. Am J Hum Genet 1996; 58: 1101–1108.
Schmidt M, du Sart D : Functional disomies of the X chromosome influence the cell selection and hence the inactivation pattern in females with balanced X; autosome translocations: a review of 122 cases. Am J Med Genet 1992; 42: 161–169.
Penaherrera MS, Ma S, Ho Yuen B, Brown CJ, Robinson WP : X-chromosome inactivation (XCI) patterns in placental tissues of a paternally derived bal t(X;20) case. Am J Med Genet 2003; 118A: 29–34.
Kleinjan DJ, van Heyningen V : Position effect in human genetic disease. Hum Mol Genet 1998; 7: 1611–1618.
Sheardown S, Norris D, Fisher A, Brockdorff N : The mouse Smcx gene exhibits developmental and tissue specific variation in degree of escape from X inactivation. Hum Mol Genet 1996; 5: 1355–1360.
Karpen GH : Position-effect variegation and the new biology of heterochromatin. Curr Opin Genet Dev 1994; 4: 281–291.
Festenstein R, Tolaini M, Corbella P et al: Locus control region function and heterochromatin-induced position effect variegation. Science 1996; 271: 1123–1125.
Milot E, Strouboulis J, Trimborn T et al: Heterochromatin effects on the frequency and duration of LCR-mediated gene transcription. Cell 1996; 87: 105–114.
Dernburg AF, Bromann KW, Fung JC et al: Pertubation of nuclear architecture by long distance chromosome interactions. Cell 1996; 85: 745–759.
Csink AK, Henikoff S : Genetic modification of heterochromatic association and nuclear organization in Drosophila. Nature 1996; 381: 529–531.
Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG : Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 1997; 91: 845–854.
Akiyama M, Kawame H, Ohashi H et al: Functional disomy for Xq26.3-qter in a boy with an unbalanced t(X.21)(q26.3;p11.2) translocation. Am J Med Genet 2001; 99: 111–114.
Acknowledgements
We thank Dr Ernestine Ngo-Piecha (Marburg) for the autopsy of the fetus, Professor Elisabeth Gödde (Recklinghausen) for the supply of parental blood, Evelyn Winkler and Daniela Lerche (Marburg) for skillful technical assistance. This work was financially supported by the PE Kempkes-Stiftung Marburg (BF, JK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fritz, B., Kunz, J., Knudsen, G. et al. Situs ambiguus in a female fetus with balanced (X;21) translocation – evidence for functional nullisomy of the ZIC3 gene?. Eur J Hum Genet 13, 34–40 (2005). https://doi.org/10.1038/sj.ejhg.5201213
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201213
Keywords
This article is cited by
-
Department für Medizinische Genetik an der Medizinischen Universität Wien
Medizinische Genetik (2007)
-
Genetics of human heterotaxias
European Journal of Human Genetics (2006)
-
Heterotaxy and cardiac defect in a girl with chromosome translocation t(X;1)(q26;p13.1) and involvement of ZIC3
European Journal of Human Genetics (2006)
