
Abstract
A late infantile metachromatic leukodystrophy patient was found to be heterozygous for the arylsulfatase A (ARSA) pseudodeficiency (pd) polyadenylation site variant (*96A>G) in the absence of the commonly associated N-glycosylation site variant (N350S). ARSA alleles were sequenced and the genotype completely defined. Six sequence variations were identified, among which two resulted as severe disease-causing mutations, both leading to the loss of the reading frame: a splice acceptor site mutation in intron 4 (849-1G>A), located on the *96A>G allele and a mononucleotide deletion (258delC) in exon 2, located on the other allele. The altered splicing caused by the 849-1G>A mutation was shown by in vitro expression of a recombinant gene containing the genomic region surrounding the mutation. Haplotype analysis of the unusual pd allele was performed in order to investigate its possible origin.
Similar content being viewed by others


Introduction
Metachromatic leukodystrophy (MLD) is an inherited disorder caused by the deficiency of the lysosomal enzyme arylsulfatase A (ARSA), which results in the accumulation of the glycolipid cerebroside sulfate in the myelin membranes of the central and peripheral nervous system, with consequent progressive demyelination.1 ARSA deficiency is due to mutations of the ARSA gene, a small gene containing eight exons that spans a genomic region of 3.2 kb in 22q13. The gene is translated into a 507 amino-acid precursor (GenBank accession number NP_000478). Onset and progression of MLD depend on the residual ARSA activity, which, in turn, is related to the severity of the mutations affecting the ARSA gene. Among the three clinical forms of MLD, the late infantile form is associated with mutations that abolish the ARSA activity, while milder mutations are present in patients affected by the other variants of the disease. Besides disease-causing mutations, variants not related to MLD have been identified in the ARSA gene. Some of these variants can slightly affect enzyme activity. A marked reduction of ARSA activity, not leading to clinical symptoms, known as ARSA pseudodeficiency (pd) is due to a pseudodeficiency allele, characterized by the presence of two variants, often in association, N350S and *96A>G, leading to the loss of a N-glycosylation site and a polyadenylation site, respectively.2 Here, we report on the molecular analysis performed on a late infantile MLD patient carrying the ARSA pd *96A>G mutation in the absence of the N350S mutation. This is a very rare condition, previously undescribed in an MLD patient.
Materials and methods
Patient and ARSA activity determination
The patient showed a clinical course and an age of onset of symptoms typical of late infantile MLD. At the age of 2 years, he showed hypotonia, regression of motor functions, and pyramidal signs. He died at the age of 6 years. ARSA activities were determined on leukocytes of the patient and his parents according to Baum et al.3
Amplification, cloning, and sequencing of the ARSA alleles of the patient
Amplification and cloning of the genomic portion of the ARSA gene containing the whole coding region was performed, as described previously.4 Clones containing the alleles from the patient were selected by PCR-RFLP for detection of the *96A>G mutation. Sequencing was performed using a 377 DNA automated sequencer with BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems). Eight primers encompassing the ARSA gene and the two vector primers M13 and M13reverse were used for sequencing. The identified mutations were confirmed by sequencing of clones obtained from different PCR products or by PCR-RFLP on genomic DNA.
Functional analysis of the 849-1G>A mutation: preparation of the pSLV1468wt and pSVL1468mut expression vectors
In order to analyze the effects of the splice site 849-1G>A mutation, a construct for in vitro expression of a recombinant gene was prepared in two versions, with or without the mutation, as described here (Figure 1). The expression vector pSVL4BAT,5 containing the ARSA cDNA, was BsaAI and BglII digested in order to eliminate the ARSA cDNA portion spanning from the 3′-end of exon 1 to the 5′-end of exon 8. At the same time, genomic DNA of the patient and of a normal control was amplified by PCR using primers SPF: 5′-GGTACGTGGGGCTGCTTGAAGAGACGCTG-3′; and SPR: 5′-GAAGATCTGGGTAGGCAGCAGGTCCAGGGAGC-3′, containing a BsaAI and a BglII site at their 5′-end, respectively. The products obtained, spanning an ARSA genomic region from the 3′-end of exon 4 to the 5′-end of exon 6, were BsaAI and BglII digested and subsequently cloned in the BsaAI/BglII-digested pSVL4BAT. Two recombinant vectors were selected, pSVL1468mut and pSVL1468wt, carrying and noncarrying the 849-1G>A mutation, respectively. In both of them the insert consisted of a recombinant ARSA gene comprising the following: the 5′-end of exon 1, followed by the genomic region spanning from the 3′-end of exon 4 to the 5′-end of exon 6, and, finally, the 3′-end of exon 8. To confirm this structure and to exclude the presence of undesired mutations, the inserts of the two vectors were sequenced using four specific primers. As expected, the two inserts differed only for the 849-1G>A mutation.

Preparation of the pSVL1468wt and pSVL1468mut expression vectors. The pSVL4BAT vector (Regis et al, 2002) was BsaAI/BglII digested cutting out almost completely the ARSA cDNA. A PCR product containing the genomic region between exons 4 and 6 of the ARSA gene was BsaAI/BglII digested and then inserted into the digested pSVL4BAT vector. The two produced pSVL1468mut and pSVL1468wt vectors differed only for the presence or absence of the 849-1G>A mutation.
Functional analysis of the 849-1G>A mutation: in vitro expression of the mutated and nonmutated recombinant gene
Transfections of the recombinant gene vectors into COS7 cells were performed, as described previously.5 Total RNA was extracted from transfected and untransfected COS cells, using the RNeasy mini kit (Qiagen). Reverse transcription was performed on total RNA with random hexanucleotides, using the SuperScript II RNase H− reverse transcriptase (Invitrogen) according to the manufacturer's protocol. Subsequent PCR was performed using primers SNF: 5′-AACCTGGACCAGCTGGCGGCGG-3′; and SNR: 5′-TGGCAGCAAGCTGGGCGGGGGG-3′, designed in exons 1 and 8, respectively, in the portions of these exons retained by the pSVL1468wt and pSVL1468mut constructs. Products were cloned in pCR2.1 (Invitrogen) and sequenced using primers M13 and M13reverse.
Results
ARSA activity of the patient was 11.1 nmol/mg/h, while the activities of the mother and the father were 56.5 and 20.2 nmol/mg/h, respectively (normal range 121±30 nmol/ mg/h).
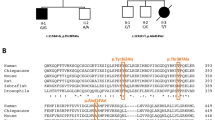
The patient was found to be heterozygous for the *96A>G mutation, but not for the N350S mutation, both commonly associated in the ARSA pd allele.2 ARSA alleles of the patient were completely sequenced. One allele was found to carry a mononucleotide deletion in exon 2 (258delC) and the common polymorphism T391S6 in exon 7. The other allele was shown to bear four nucleotide substitutions: a mutation leading to the loss of the acceptor splice site in intron 4 (849-1G>A), the W193C and T391S polymorphisms,6 in exons 3 and 7, respectively, and the *96A>G pd variant in the first polyadenylation site (Figure 2).

ARSA alleles of the patient. The disease-causing mutations are indicated by squares, the variants by ovals.
While T391S, W193C and *96A>G are known variants whose role is largely documented,2,6 258delC and 849-1G>A are new mutations.
The 258delC mononucleotide deletion causes a frameshift at codon 86 of the wild-type ARSA mRNA (GenBank accession number NM_000478), leading to a predicted truncated protein of 104 amino acids.
As regards the 849-1G>A mutation, since RNA of the patient for the analysis of the mutated transcript was not available, we set up a simple system for in vitro expression of a recombinant gene containing the genomic region surrounding the mutation. The gene consisted of a deleted ARSA cDNA in which a fragment of ARSA genomic DNA was inserted (Figure 3). COS cells were transfected with two expression vectors, pSVL1468wt, containing the nonmutated recombinant gene, and pSVL1468mut, containing the mutated version of the gene. The expression of the recombinant gene was checked by RT-PCR using primers flanking the gene. An RT-PCR product of 311 bp was obtained from COS cells transfected with pSVL1468wt, while COS cells transfected with pSVL1468mut yielded two shorter RT-PCR products of 271 and 186 bp, respectively, in an apparently equivalent amount. No product was obtained from untransfected COS cells. Sequence analysis of the 311 bp product confirmed the regular splicing of the nonmutated recombinant gene. On the other hand, the sequence of the 271 bp product showed the incorrect splicing of the gene due to the use of a cryptic acceptor splice site in the 5′-portion of intron 5 in place of the mutated acceptor splice site in intron 4. Consequently, exon 5 was completely lost, while 85 out of 90 nucleotides of intron 5 were transcribed. The 186 bp product showed the skipping of exon 5. All the splice junctions conformed to the AG/GT rule.

The recombinant gene used for the expression experiments. The splicing of the gene was analyzed by RT-PCR performed on COS cells transfected with pSVL1468wt (lane wt), with pSVL1468mut (lane mt), or untransfected (lane un). The nonmutated version of the gene was regularly spliced by COS cells transfected with pSVL1468wt, as confirmed by the sequencing of the 311 bp RT-PCR product. The mutated version of the recombinant gene was incorrectly spliced by COS cells transfected with pSVL1468mut in two different ways, as indicated by the sequencing of the 271 and 186 bp RT-PCR products. The portion of intron 5 nonspliced in the 271 bp splice variant is reported. The cryptic acceptor splice site used in place of the mutated intron 4 acceptor site is underlined. The molecular weight marker (Φ) is ΦX 174 DNA HaeIII digested.
Both altered modes of splicing due to the 849-1G>A mutation cause a frameshift at codon 283 of the ARSA mRNA (GenBank accession number NM_000478), leading to the production of truncated proteins of 304 and 314 amino acids, respectively.
Using data obtained by sequencing of the allele bearing the polyadenylation site variant, we were able to define its haplotype. Three polymorphisms were considered, according to Zlotogora et al.7 The allele showed the absence of the BglI site in exon 3 (the presence of the W193C variant), of the BsrI site in exon 7 (presence of the T391S variant), and of the BamHI site in intron 7.
The inheritance of the disease alleles was easily established by PCR-RFLP methods: the 258delC-containing allele was inherited from the mother, while the 849-1G>A-containing allele was inherited from the father. Moreover, the ARSA allele of the father not transmitted to the patient was a classic pd allele, carrying the N350S and *96A>G variants.
Discussion
Among the nucleotide variations identified in the ARSA alleles of the patient, the two novel mutations were shown to be severe disease-causing mutations, leading to the production of truncated ARSA proteins. While the 258delC mutation evidently causes a frameshift in the ARSA gene, the effects of the 849-1G>A mutation were shown by the use of an in vitro system, since RNA of the patient was not available. The system is very simple, consisting of COS7 cells transfected with an expression vector containing the desired gene. The splicing of the gene, in the presence or absence of the mutation, was easily analyzed. Moreover, the use of a recombinant gene allowed a clear discrimination between the expression of the target gene and the expression of the corresponding endogenous gene of the COS cells.
As seen before, besides the two described mutations, the alleles of the patient were found to carry other nucleotide variants. The T391S mutation, which was identified in both the alleles, can cause a limited reduction of the ARSA activity (about 11%).5 The W193C mutation, contained in the 849-1G>A allele, does not change the ARSA activity with respect to the wild type.6 The *96A>G mutation, also located on the 849-1G>A allele, is reported to cause a severe deficiency of the 2.1 kb ARSA mRNA species, which represents 90% of the poly(A)+ ARSA mRNA, according to Gieselmann et al2 or 71% of the total available ARSA mRNA, according to Harvey et al.8 Therefore, the transcript containing the 849-1G>A mutation was presumably much more infrequent than the other transcript in vivo. On the other hand, the reported variants had no effect on the enzyme activity of the patient, being located on ARSA alleles containing null mutations. Likely, none of these variants played a role in affecting the clinical phenotype. Anyway, the presence of variants like T391S and *96A>G, which can reduce the ARSA activity, should be taken into account when occurring on the allele background of mild disease-causing mutations. In these situations, they can potentially contribute to disease severity.5
Interestingly, the *96A>G mutation was found not associated, as usual, with the N350S mutation in the typical pd allele.2 The two mutations are often found together, but, not rarely, the N350S mutation is found alone.9 On the other hand, the *96A>G mutation has been previously found not linked to the N350S mutation only in a very limited number of cases: a subject reported by Leistner et al,10 two sibs reported by Ricketts et al,11 and two unrelated subjects reported by Gort et al.12 None of these subjects was an MLD patient. This is the first reported case in which the unusual pd allele containing the polyadenylation site variant but lacking the N350S variant bears a disease-causing mutation.
As seen in the results, the haplotype of this unusual N350S−/*96A>G+/849-1G>A+ pd allele was analyzed according to Zlotogora et al,7 finding the absence of the three polymorphic BglI, BsrI, and BamHI sites. The pd allele reported by Ricketts et al11 did not carry the BsrI and BamHI sites, but carried the BglI site. The classic pd haplotype reported by Zlotogora et al7 contained the BglI and BsrI sites, but not the BamHI site. Therefore, the N350S−/*96A>G+/849-1G>A+ pd allele clearly differs from both Ricketts' pd allele and the classic pd allele. We can argue that the two unusual pd alleles are unrelated to each other, and, moreover, that the N350S−/*96A>G+/849-1G>A+ pd allele did not derive from the classic pd allele. Consequently, it is not possible to speculate about the origin of the N350S−/*96A>G+/849-1G>A+ pd allele, in which the 849-1G>A mutation could have occurred before or after the occurrence of the *96A>G mutation.
A good correlation was found between patient's and parents' genotypes and the corresponding ARSA activity data. According to molecular and biochemical data, the proband was a typical late infantile MLD patient, carrying two MLD-causing ARSA alleles that were responsible for a virtually absent enzyme activity. The mother was a common ARSA heterozygote, with an enzyme activity that was about half the normal due to the presence of a wild-type and an MLD allele. A case apart was represented by the father, who showed a very low residual ARSA activity due to the disease allele transmitted to the patient and to a classic pd allele. He was therefore an MLD/pd compound heterozygote. This is a particular genotype, which is associated with a residual ARSA activity that is lower than in a healthy pd homozygote but higher than in an adult MLD patient. MLD/pd heterozygotes are usually healthy, as the father of the patient was at the time of the diagnosis. However, some cases of MLD/pd subjects with nonprogressive neurological symptoms have been reported.4,13 It is still unclear whether this fact is coincidental or the MLD/pd compound heterozygosity predisposes to neurological disorders in the presence of unknown genetic or nongenetic factors.
References
von Figura K, Gieselmann V, Jaeken J : Metachromatic leukodystrophy; in Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 2001, pp 3695–3724.
Gieselmann V, Polten A, Kreysing J, von Figura K : Arylsulfatase A pseudodeficiency: loss of polyadenylation signal and N-glycosylation site. Proc Natl Acad Sci USA 1989; 86: 9436–9440.
Baum H, Dogson KS, Spencer B : The assay of the arylsulfatase A and B in human urine. Clin Chim Acta 1959; 4: 453–455.
Regis S, Filocamo M, Stroppiano M, Corsolini F, Caroli F, Gatti R : A 9bp deletion on the background of the arylsulfatase A pseudodeficiency allele in a late infantile metachromatic leukodystrophy patient and in a patient with nonprogressive neurological symptoms. Hum Genet 1998; 102: 50–53.
Regis S, Corsolini F, Stroppiano M, Cusano R, Filocamo M : Contribution of arylsulfatase A mutations located on the same allele to enzyme activity reduction and metachromatic leukodystrophy severity. Hum Genet 2002; 110: 351–355.
Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V : Molecular basis of different forms of metachromatic leukodystrophy. N Engl J Med 1991; 324: 18–22.
Zlotogora J, Furman-Shaharabani Y, Goldenfum S, Winchester B, von Figura K, Gieselmann V : Arylsulfatase A pseudodeficiency: a common polymorphism which is associated with a unique haplotype. Am J Med Genet 1994; 52: 146–150.
Harvey JS, Carey WF, Phillip Morris C : Importance of the glycosylation and polyadenylation variants in metachromatic leukodystrophy pseudodeficiency phenotype. Hum Mol Genet 1998; 7: 1215–1219.
Ott R, Waye JS, Chang PL : Evolutionary origins of two tightly linked mutations in arylsulfatase-A pseudodeficiency. Hum Genet 1997; 101: 135–140.
Leistner S, Young E, Meaney C, Winchester B : Pseudodeficiency of arylsulphatase A: strategy for clarification of genotype in families of subjects with low ASA activity and neurological symptoms. J Inherit Metab Dis 1995; 18: 710–716.
Ricketts MH, Goldman D, Long JC, Manowitz P : Arylsulfatase A pseudodeficiency-associated mutations: population studies and identification of a novel haplotype. Am J Med Genet 1996; 67: 387–392.
Gort L, Coll MJ, Chabas A : Identification of 12 novel mutations and two new polymorphisms in the arylsulfatase A gene: haplotype and genotype–phenotype correlation studies in Spanish metachromatic leukodystrophy patients. Hum Mutat 1999; 14: 240–248.
Penzien JM, Kappler J, Herschkowitz N et al: Compound heterozygosity for metachromatic leukodystrophy and arylsulfatase A pseudodeficiency alleles is not associated with progressive neurological disease. Am J Hum Genet 1993; 52: 557–564.
Acknowledgements
The DNA samples used were obtained by the collection ‘Cell Line and DNA Bank from Patients affected by Genetic Disease’ (www.gaslini.org/labdppm.htm) supported by Italian Telethon grants (Project No. GTF02012).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Regis, S., Corsolini, F., Ricci, V. et al. An unusual arylsulfatase A pseudodeficiency allele carrying a splice site mutation in a metachromatic leukodystrophy patient. Eur J Hum Genet 12, 150–154 (2004). https://doi.org/10.1038/sj.ejhg.5201100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201100
Keywords
This article is cited by
-
Clinical, Biochemical, and Molecular Characterization of Metachromatic Leukodystrophy Among Egyptian Pediatric Patients: Expansion of the ARSA Mutational Spectrum
Journal of Molecular Neuroscience (2021)
-
An Italian Cohort Study Identifies Four New Pathologic Mutations in the ARSA Gene
Journal of Molecular Neuroscience (2013)
