
Abstract
Mutations in spastin cause the most common form of pure autosomal dominant hereditary spastic paraparesis (SPG4). Here, we report two Italian families affected with SPG4-linked HSP harboring two novel spastin mutations. SSCP/sequencing analysis of the spastin gene showed a single base pair deletion causing a frame-shift in one family (1442delT) and a missense mutation (1726T>C) resulting in a leucine to proline amino-acid change (L534P) in the other family. Total RNA from the mutant and the wild-type spastin allele in muscle biopsies from patients from the two affected families was quantitated. RNA expression was almost absent from the spastin allele harboring the single base pair deletion, while it was nearly normal for the spastin allele harboring the missense mutation. These data suggest that varying spastin RNA levels are found in out-of-frame and missense spastin mutations and imply different mechanisms involved in the molecular pathology of SPG4 linked HSP.
Similar content being viewed by others

Introduction
Autosomal dominant hereditary spastic paraparesis (AD-HSP) linked to chromosome 2p (SPG4) is the most common form of AD-HSP. Mutations in the SPG4 gene, spastin, result in a pure form of spastic paraparesis characterized by slow and progressive weakness and bilateral spasticity of the lower limbs.1
Missense, nonsense, frameshift and splice-site mutations have been reported in SPG4, but correlation between any of these mutations and clinical severity has not been identified.1,2
Despite the large variety of mutations of the SPG4 gene reported, the molecular mechanisms underlying SPG4-linked HSP are still unclear. SPG4 transcript, spastin, is ubiquitously expressed in adult and fetal tissues.1 It has been suggested that haploinsufficiency is the molecular cause of the disease since most of the missense mutations reported in the SPG4 gene are located in the AAA cassette altering the functional domain of the protein, while other mutations, scattered along the entire coding sequence and leading to premature stop codons, result in RNA instability.1,3 Recently it has been shown that spastin protein interacts dynamically with microtubules in transfected Cos cells.4 Expression of missense mutations located in the AAA domain resulted in a redistribution of the microtubule cytoskeleton. This was the first evidence that a dominant-negative mechanism could be pathogenic in patients with spastin missense mutations.4
Here we present spastin RNA studies in two Italian AD-HSP families segregating two novel SPG4 mutations to start to dissect the molecular pathogenesis of SPG4-linked HSP.
Material and methods
Family data
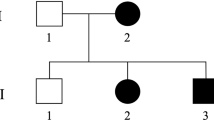
We studied three affected individuals from two Italian families (004 and 009) segregating a pure form of an autosomal dominant HSP. Clinical symptoms in affected individuals in both families included spastic paraparesis with exaggerated tendon reflexes in the lower limbs, Babinski sign, decreased vibration sense in the lower limbs, pes cavus and mild urinary urgency in two cases. A muscle biopsy was performed in two individuals in family 004, and in one in family 009 after informed consent.
Spastin gene mutation studies
DNA was isolated from peripheral blood. A total of 26 sets of primers were designed to amplify all spastin exons (primer sequences available upon request). Three different single-strand conformational polymorphism (SSCP) conditions were used for screening for SPG4 gene mutations. Conformers were reamplified using the original amplification primer and PCR conditions, and directly sequenced on an automatic sequencer.
The identified spastin mutations were confirmed with appropriate PCR primers designed to cover the two mutated regions (primer sequences available upon request). For family 004, the PCR products were size-fractioned in denaturing acrylamide gel electrophoresis and for family 009, where the mutation causes a loss of BfaI restriction endonuclease site, restriction fragment length analysis was done.
Quantitation of spastin RNA by quantitative multiplex fluorescent RT-PCR (QMF-RT/PCR)
A muscle biopsy was obtained from two individuals in family 004 and from one individual in family 009. As controls, muscle biopsies from eight patients who underwent orthopedic surgery for unrelated disorders were studied. We designed two fluorescent primer sets for coamplification of cDNAs corresponding to laminin α2 (LAMA2) and spastin gene (SPG4). Spastin primer sets were designed to encompass the mutated region in order to distinguish the wild-type and mutated spastin allele on the basis of their molecular weight. Fluorescent RT-PCR products were loaded on an ABI 373 DNA Analyzer. Peak areas corresponding to the mutated and wild-type SPG4 allele and the LAMA2 internal control were determined. Quantitation of fluorescent PCR products of SPG4 relative to LAMA2 was done as previously described.5
Results
Spastin mutation studies
With exon 10 primers, an aberrant conformer was detected in both individuals in family 004 and with exon 14 primers in an affected individual in family 009. Direct sequencing of the conformers showed a single base pair deletion at position 1442 of the spastin coding sequence (1442delT), resulting in a stop codon at position 1443 in family 004 and a T to C nucleotide change at position 1726 (1726 T>C) in family 009. The 1726 T>C mutation resulted in a leucine to proline amino-acid change at residue 534 (L534P).
To confirm the identified spastin mutation in family 004, a PCR primer set was designed encompassing the deletion. PCR products were size-fractioned on acrylamide gel and a heterozygous mobility shift band was identified in both patients (Figure 1, panel a). The 1726T>C mutation in family 009 causes a loss of a BfaI restriction endonuclease site.

Identification of two novel spastin mutations in SPG4 families 004 (a) and 009 (b). Panel A shows a maternally inherited single base pair deletion in patients II-1 (C are controls). Shown is autoradiography of the PCR products encompassing the one base pair deletion at nucleotide position 1442 of the spastin coding sequence resulting in a stop codon at nucleotide position 1443. I-1 and II-1 are heterozygous for the normal size and for the deleted allele. Panel b shows the maternal inheritance of a spastin missense mutation (1726T>C) in patient III-1 (C are controls). The 1726T>C mutation causes a loss of BfaI restriction enzyme site. The autoradiography of the digested PCR product encompassing the 1726T>C mutation is shown. II-1 and III-1 are heterozygous for the mutation. Dashes show the three fragments resulting from BfaI enzymatic digestion in the controls (160, 39 and 13 bp). In the patients the loss of a BfaI site resulted in four fragment-sized 173, 160, 39 and 13 bp.
Appropriate PCR products were BfaI digested and the restriction fragment length analysis showed that the affected individual harbors the 1726T>C mutation (Figure 1, panel b), absent from 100 normal chromosomes.
Quantification of spastin RNA in muscle biopsy
RNA was isolated from muscle biopsy of two individuals in family 004, from one in family 009 and from eight controls. RNA quantitation was done for both the wild type and the mutated spastin allele in families 004 and 009. The single base pair deletion identified in family 004 (1442delT) resulted in a spastin mutated allele one base pair shorter (101 bp) than wild type (102 bp) (Figure 2, panel a); in family 009 the 1726T>C nucleotide change causes a loss of BfaI restriction site. BfaI-digested PCR products resulted in a single PCR product in controls, corresponding to the wild-type allele (116 bp), and in two PCR products in family 009 corresponding to the wild (116 bp) and mutated allele (129 bp) (Figure 2, panel b). Peak areas and the ratio between SPG4 vs LAMA2 were calculated for the patients and the control group. The average SPG4/LAMA2 ratio in the controls was set to 100%, and each single value and the average SPG4/LAMA2 ratio in families 004 and 009 normalized to this (Figure 3, panels a and b). Patients from family 004 showed a marked decrease in spastin RNA relative to the mutated SPG4 allele (4% of the control). The wild-type SPG4 allele was expressed at about 66% of the control level. When both wild and mutated SPG4 alleles were considered together and compared to controls, the RNA was still significantly decreased (70%). Patient from family 009 showed 81% of total SPG4 RNA level (wild and mutated type) seen in controls. SPG4 mutated allele was at about 45% of controls and the normal SPG4 allele at 36%.

Quantitative multiplex fluorescent analysis of spastin RNA. Shown are the unprocessed electrophoretograms of 22 cycles fluorescent RT-PCR products from patient II-1 from family 004 in panel a, and from patient III-1 from family 009 in panel b. Spastin mutation identified in SPG4 family 004 (panel a) is a single-nucleotide deletion at position 1442 (1442delT) of the spastin coding sequence. PCR products encompassing the deletion result in two peaks in the patient traces, corresponding to the wild-type (SPG4 in the figure) and the mutated (del1442T SPG4 in the figure) spastin allele. Quantitation of RNA showed a marked decrease of mutated spastin allele RNA in the patient. Spastin mutation identified in family 009 (b) is a T to C nucleotide change at position 1726 of the spastin coding sequence resulting in a loss of a BfaI restriction enzyme site. In panel b, BfaI-digested PCR products result in two peaks in the patient traces and correspond to the wild (SPG4) and mutated (T1726C SPG4) spastin allele. Quantitation of RNA showed a normal amount of RNA from the spastin allele harboring the missense mutation and from the wild-type allele.

Quantitation of spastin RNA levels in the SPG4 patients' muscle biopsies relative to laminin α2 (LAMA2) in total RNA pools. Shown are the results of multiple fluorescent PCR assays using cDNA from two individuals of family 004, from one individual of family 009 and from eight control muscle biopsies. Each data point shows an independent RT-PCR reaction and subsequent peak area quantitation using an automated sequencer. SPG4/LAMA2 ratios were calculated for each patient and control. The average and standard deviation relative to the normal, mutated, and to the sum of both in SPG4 families and to total SPG4 RNA in the controls is shown to the right of each set of values with a filled triangle. Individuals from family 004 show a decrease of spastin RNA from the mutated spastin allele (4% ± 2.6%) resulting in a decreased total SPG4 RNA (a), where individual from family 009 did not show any decrease of spastin RNA from the mutated spastin allele (45%±2.6%) thus not affecting total SPG4 RNA (b).
Discussion
We performed spastin RNA studies in two AD-HSP families segregating two novel SPG4 mutations. Haploinsufficiency has been suggested as a possible mechanism in SPG4 since most of the spastin mutations result either in an unstable aberrant transcript,1,3 or in a decreased number of functional proteins due to inactivation of functional spastin domains caused by spastin missense mutations. Recently, RNA and protein studies in two nonsense and one frameshift mutation in lymphoblastoid cell lines of HSP patients showed a reduced amount of spastin, in comparison with one missense mutation that resulted in a normal amount of the protein,3 however, no data were provided on the expression level from the spastin mutated allele.
To dissect the molecular pathogenesis of SPG4, we conducted a series of experiments measuring the amount of spastin RNA from both the normal and mutated spastin alleles in the muscle biopsy of three patients affected with SPG4-linked HSP. Since spastin is ubiquitously expressed,1 and muscle is a very accessible tissue, we chose to use skeletal muscle biopsy as the RNA source; however, it is important to note that our data on skeletal muscle may or may be not reflect SPG4 transcriptional activity in the neurons affected by HSP. Our results suggest that the pathogenetic mechanisms in nonsense/frameshift and missense spastin mutations in SPG4 are different at the RNA level and therefore possibly at the translational level, even if a combined effect of transcriptional activity and RNA degradation may both contribute to RNA aboundance. While in nonsense/frameshift spastin mutation a dosage effect is well established, in missense mutation both a dominant-negative mechanism and/or a functional inactivation of the protein are likely, although a haploinsufficiency mechanism cannot be completely excluded. Interestingly, it has been suggested by in vitro studies of the effects of different spastin missense mutations in microtubules dynamics that mutated spastin binds constitutively to microtubules, leading to a redistribution of the microtubule cytoskeleton.4 These data strongly suggest that the mutated spastin protein can hamper the spastin function through a dominant-negative effect.
References
Hazan J, Fonknechten N, Mavel D et al: Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet 1999; 23: 296–303.
Fonknechten N, Mavel D, Byrne P et al: Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet 2000; 9: 637–644.
Charvin D, Cifuentes-Diaz C, Fonknechten N et al: Mutations of SPG4 are responsible for a loss of function of spastin, an abundant neuronal protein localized in the nucleus. Hum Mol Genet 2003; 12: 71–78.
Errico A, Ballabio A, Rugarli EI: Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet 2002; 11: 153–156.
Morrone A, Pegoraro E, Angelini C, Zammarchi C, Marconi G, Hoffman EP: RNA metabolism in myotonic dystrophy. Patient muscle shows decreased insulin receptor RNA and protein consistent with abnormal insulin resistance. J Clin Invest 1997; 99: 1691–1698.
Acknowledgements
We thank the family members for their cooperation in this study. This work was Supported by a University of Padova grant (‘Fondi Destro’), a University of Padova grant for Young Investigators (AM), a Thelethon grant (TF0003Y01) and MURST 2001068328001.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Molon, A., Montagna, P., Angelini, C. et al. Novel spastin mutations and their expression analysis in two Italian families. Eur J Hum Genet 11, 710–713 (2003). https://doi.org/10.1038/sj.ejhg.5201027
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201027
