
Abstract
Hereditary cataract is a clinically and genetically heterogeneous lens disease that accounts for a significant proportion of visual impairment and blindness in childhood. The alphaA-crystallin (CRYAA) gene (CRYAA) encodes a member of the small-heat-shock protein (sHSP) family of molecular chaperones and is primarily and abundantly expressed in the ocular lens. Here, we have used linkage analysis to identify a novel missense mutation in CRYAA that underlies an autosomal dominant form of ‘nuclear’ cataract segregating in a four-generation Caucasian family. A maximum two-point LOD score (Zmax) of 2.19 (maximum recombination fraction, θmax=0) and multipoint Zmax of 3.3 (θmax=0) was obtained at marker D21S1885. Haplotype analysis indicated that the disease gene lay in the ∼2.7 Mb physical interval between D21S1912 and D21S1260 flanking CRYAA on 21q22.3. Sequence analysis identified a C → T transition in exon 1 of CRYAA from affected individuals that was predicted to result in the nonconservative substitution of cysteine for arginine at codon 49 (R49C). Transfection studies of lens epithelial cells revealed that, unlike wild-type CRYAA, the R49C mutant protein was abnormally localized to the nucleus and failed to protect from staurosporine-induced apoptotic cell death. This study has identified the first dominant cataract mutation in CRYAA located outside the phylogenetically conserved ‘alpha-crystallin core domain’ of the sHSP family.
Similar content being viewed by others

Introduction
Congenital, infantile and childhood forms of cataract present with an estimated incidence of 2.5–3.5 cases per 10 000 live births,1 and account for ∼4% of adult blindness in industrialized countries.2 At least one-third of all cases may have a genetic basis,3 either as part of a multisystem disease or, more frequently, as a nonsyndromic Mendelian trait.4 All three classical forms of Mendelian inheritance have been reported, with the majority of cases exhibiting autosomal dominant transmission. To date at least 20 independent loci for clinically diverse forms of Mendelian cataract have been mapped on 14 human chromosomes.4 No causative genes have been reported at the dominant loci on chromosomes 1p,5,6 2p,7 15q,8 17p,9 17q2410 and 20p11 or at the recessive loci on 3p12 and 9q.13 However, underlying mutations have been identified in seven crystallin genes including: the alpha-crystallin genes located on 11q (CRYAB)14 and 21q (CRYAA),15,16 three beta-crystallin genes located on 17q (CRYBA3/A1)17,18 and 22q (CRYBB2, CRYBB1)19,20,21,22 and two gamma-crystallin genes (CRYGC, CRYGD) located on 2q.23,24,25,26 In addition, five genes for lens membrane/cytoskeletal proteins including those for connexin50 (GJA8) on 1q,27,28,29 connexin46 (GJA3) on 13q,30,31 aquaporin-0 (MIP) on 12q,32 a peripheral myelin-like protein (LIM2) on 19q33 and an intermediate filament-like protein (BFSP2) on 3q34,35 have been linked with Mendelian cataract. Moreover, certain mutations in genes widely expressed outside the lens, including those for paired-like homeodomain transcription factor 3 (PITX3) on 10q,36 heat-shock transcription factor 4 (HSF4) on 16q,37 galactokinase 1 (GALK1) on 17q38 and L-ferritin (FTL) on 19q,39 have been associated with cataract in the absence of other significant ocular or systemic defects. To gain further insight about the pathogenetic complexity of hereditary cataract, we have carried out linkage analysis in a family segregating autosomal dominant ‘nuclear’ cataract and subsequently identified a novel missense mutation in the gene for alphaA-crystallin (CRYAA) on 21q, which encodes a member of the small-heat-shock protein (sHSP) family of molecular chaperones.40
Materials and methods
Genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood leukocytes using the QIAamp DNA blood maxi kit (Qiagen) according to the manufacturer's instructions. Généthon microsatellite (CA)n repeat markers41 were amplified using the polymerase chain reaction (PCR). Each reaction (10 μl) contained 10 ng of genomic DNA, 1 × PCR buffer, 1.5 mM MgCl2, 250 μ M dNTPs, 0.15 pmol each of sense and antisense primers and 0.05 U of Taq DNA polymerase (Gibco). All sense primers were 5′ end-labeled with infrared dye 800 (Li-Cor). Thermal cycling was performed using a DNA engine (MJ Research) and a touch-down protocol recommended by Li-Cor. Fluorescently labeled PCR products were separated on a 6.5% acrylamide gel and detected using a Li-Cor 4200 DNA analyzer running Gene ImageR software (Li-Cor). Pedigree and haploptype data were managed using Cyrillic (2.1) software (Family Genetix Ltd.). Two-point and multipoint LOD scores (Z) were calculated using the MLINK and LINKMAP subprograms, respectively, from the LINKAGE (5.1) package of programs.42 Microsatellite marker allele frequencies used for linkage analysis were those calculated by Généthon.41 A gene frequency of 0.0001 and a penetrance of 100% were assumed for the cataract locus.
Mutation analysis
Consensus exon/intron boundaries in CRYAA were verified by aligning the appropriate cDNA sequence43 with the corresponding genomic sequence44 assigned to chromosome 21 (BAC clone 445C9). Gene-specific primers (Table 1) were designed to anneal to intronic sequence flanking exon boundaries. Genomic DNA (100 ng) was PCR amplified in a 50 μl reaction containing 1 × PCR buffer, 1.5 mM MgCl2, 250 μ M dNTPs and 1.25 U Tag polymerase (Gibco) and 25 pmols of each appropriate intron-specific primer (Table 1) using a DNA engine (MJ Research). PCR products were purified by QIAquick gel extraction kit (Qiagen) and sequenced in both directions using dichloro-rhodamine terminator chemistry on an ABI 310 DNA sequencer (Applied Biosystems). Restriction fragment length analysis was performed using Aci1 (New England Biolabs) according to the manufacturer's instructions. Digestion products were then separated and visualized in 2% agarose gels containing 0.05% ethidium bromide. In order to distinguish the predicted mutation, with 95% confidence, from a polymorphism with 1% frequency, we carried out Aci1 restriction analysis or direct sequencing analysis of genomic DNA samples from a panel of 170 unrelated control individuals as recommended previously.45
Site-directed mutagenesis
The cDNA sequence encoding wild-type human CRYAA was subcloned into the pCIneo mammalian expression vector (Promega) as described previously.46 Site-directed mutagenesis of wild-type CRYAA to R49C-CRYAA was performed using the QuikChange Mutagenesis kit (Stratagene) according to the manufacturer's recommendations and verified by sequencing prior to transfection. Sequences of sense and antisense primers used to introduce the C → T mutation at nucleotide 145 of the CRYAA coding region were; 5′ CC ATC AGC CCC TAC TAC TGC CAG TCC CTC T TCC GC and 5′ GCG GAA GAG GGA CTG GCA GTA GTA GGG GCT GAT GG, respectively.
Cell culture and transfection
Human lens epithelial cells with extended lifespan (HLE B-3) were passaged using Trypsin-EDTA, plated on 35 mm dishes and cultured in Eagle's minimum essential medium supplemented with 20% fetal bovine serum and gentamicin (50 μg/ml) as described previously.46,47 DNA transfection was performed by calcium phosphate precipitation followed by Me2SO-shock treatment using the ProFection kit (Promega) according to the manufacturer's recommendations then geneticin (0.5 mg/ml)-resistant colonies were selected and expanded as described previously.46,47 Clonal transfectants were used for up to four passages without detectable changes in the levels of exogenous wild-type or mutant CRYAA as determined by immunoblot analysis.
Immunoblot analysis
Quantitative immunoblot analysis was performed as described previously.46,47 Briefly, cell lysates were prepared from transfected cells (∼106), resolved on SDS-polyacrylamide (12%) gels, blotted onto Immobilon membranes (Millipore) and immunolabeled with a polyclonal antiserum to bovine alpha-crystallin (diluted 1:1000) followed by 125I-protein A (Amersham) and imaged using a Storm 860 phosphoimaging system running ImageQuant software (Molecular Dynamics).
Immunofluorescence microscopy
Transfected cells, plated on sterile glass coverslips in 35 mm dishes, were fixed (30 min) with 4% paraformaldehyde, permeabilized (30 min) in 0.1% Triton X-100, blocked (30 min) in 10% normal goat serum, and then incubated (4°C) with appropriate primary (16 h) and secondary (1 h) antibodies as described previously.46,47 Briefly, to detect CRYAA, cells were immunolabeled with a monoclonal antibody (1:50 dilution) to bovine alphaA-crystallin (a gift from Dr Paul Fitzgerald) followed by an Alexa568-conjugated goat anti-mouse IgG (Molecular Probes). To detect CRYAB, cells were immunolabeled with an antibody (1:100 dilution) to bovine alphaB-crystallin (Nova Castra) followed by an Alexa488-conjugated goat anti-rabbit IgG (Molecular probes). To visualize the actin cytoskeleton, in the absence of CRYAB immunolabeling, cells were incubated (20 min) in fluorescein–phalloidin (2 U/ml PBS, Molecular Probes), then washed in PBS (3 × 5 min) and viewed under an LSM 410 confocal microscope equipped with an argon–krypton laser (Zeiss).
Cryoimmunoelectron microscopy
Transfected cells were trypsinized and fixed in 4% parafomaldehyde, embedded in gelatin, and infiltrated with a cryopreservative, consisting of 2.3 M sucrose/10% polyvinylpyrrolidone, at 4°C. Samples were frozen in liquid nitrogen and sectioned (50–80 nm) using a cryoultramicrotome (Reichert), then blocked and incubated with the appropriate primary and secondary antibodies using standard techniques. The primary antibody to CRYAA was the same as that used for immunofluorescence above, and a goat anti-mouse IgG conjugated with 18 nm gold particles was used as the secondary antibody (Sigma). Specimens were stained with uranyl acetate and viewed under a 1200EX transmission electron microscope (JEOL).
Fluorescence-activated cell sorting (FACS) analysis
Transfected cells were treated with staurosporine (0.5 μ M, 2 h), then labeled with Annexin V-FITC and propidium iodide (PI) (Pharmingen), according to the manufacturer's instructions, and the resulting apoptotic cells quantified by flow cytometry using a FACScan running CellQuest software (Becton Dickinson) as described previously.47
Results
Linkage to CRYAA on 21q
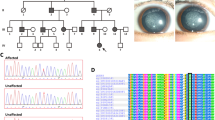
We ascertained a four-generation Caucasian family segregating autosomal dominant ‘nuclear’ cataract in the absence of other ocular or systemic abnormalities (Figure 1). Ophthalmic records indicated that the bilateral opacities presented at birth or during infancy and were confined to the central zone or fetal nucleus of the lens. Following informed consent, 14 members of the family (Figure 1), including 10 affected individuals, three unaffected individuals and one spouse, were genotyped with microsatellite markers from the (CA)n map41 at two crystallin loci linked with autosomal dominant nuclear cataract on 2q32–q36 (CRYGD) and 21q22.3 (CRYAA). After exclusion of CRYGD with D2S128 (Z=−2.32, θ=0.05), we obtained suggestive evidence of linkage (Table 2) for marker D21S1885 (Z=2.19, θ=0). Multipoint analysis using markers D21S1260, D21S1885 and D21S1912 yielded a Zmax of 3.30 (θmax=0) at D21S1885 (data not shown).

Pedigree and haplotype analysis of the cataract family showing segregation of five microsatellite markers on chromosome 21q, listed in descending order from the centromere. Squares and circles symbolize males and females, respectively. Filled symbols denote affected status.
Haplotype analysis of the pedigree (Figure 1), detected seven affected individuals (II:8, III:3, III:5, IV:1, IV:3, IV:4, IV:5) and one unaffected female (III:6) that were recombinant at D21S1260. It is likely that six of the affected individuals (III:3, III:5, IV:1, IV:3, IV:4, IV:5) inherited the same recombination event that originated independently in their deceased ancestor II:5. In addition, two affected relatives (III:3, IV:1) were recombinant at D21S1912. However, no individuals recombinant for the cataract locus and D21S1885 were detected, indicating that the disease gene lay in the physical interval D21S1260-(2.0 Mb)-D21S1885-(0.7 Mb)-D21S1912. According to the chromosome 21 database (www.ensembl.org), D21S1885 lies ∼0.25 Mb distal to CRYAA, suggesting that the latter was a strong candidate gene for the cataract.
CRYAA mutation analysis
The CRYAA gene comprises three coding exons and two introns15 (Figure 2d). Sequence analysis of all three exons and immediate flanking regions in two affected individuals using intron-specific primers (Table 1) detected a heterozygous C → T transition in exon 1 that was not present in wild type (Figure 2). This single-nucleotide change deleted one of three Aci1 restriction sites present in exon 1. Restriction fragment length analysis (Figure 2c) confirmed the presence of the heterozygous C → T transition in all affected members of the pedigree and its absence in three unaffected relatives (II:3, III:6, IV:2) and one spouse (II:4). Furthermore, when we tested the C → T transition as a biallelic marker, with a notional allelic frequency of 1%, in a two-point LOD score analysis of the cataract locus (Table 2), we obtained significant evidence of linkage (Z=3.59, θ=0). Finally, we excluded the C → T transition as a single-nucleotide polymorphism in a panel of 170 normal unrelated individuals (data not shown). At the level of protein translation, the C → T transition was predicted to result in a missense substitution of arginine to cysteine at codon 49 (R49C). Alignment of amino-acid sequences for CRYAA present in the Entrez Protein database (ncbi.nlm. nih.gov/Entrez/) revealed that arginine 49 is phylogenetically conserved from zebrafish to man (data not shown). Moreover, the predicted R49C substitution represented a nonconservative amino-acid change, with the positively charged (basic) polar side group of arginine replaced by the uncharged polar sulfydryl side group of cysteine. Taken overall, the cosegregation of the C → T transition only with affected members of the pedigree and its absence in 340 normal chromosomes strongly suggested that the nonconservative R49C substitution was a causative mutation rather than a benign polymorphism in linkage disequilibrium with the disease.

Mutation analysis of CRYAA. Sequence chromatograms of wild-type allele (a) showing translation of arginine (CGC) at codon 49 and mutant allele (b) showing a C → T transition in exon 1 at the first base of codon 49 that substituted cysteine (TGC) for arginine (R49C). (c) Restriction fragment length analysis showing loss of an Aci1 site (5′C ↓ CGC) that cosegregated with affected individuals heterozygous for the R49C mutation (206 and 191 bp alleles), but not with unaffected individuals (191 bp allele). (d) Exon organization and mutation profile of CRYAA. Codon numbers are shown above each exon. The conserved ‘alpha-crystallin’ domain is shaded gray. The relative locations of the R49C mutation and four other mutations associated with cataract in humans (W9X, R116C) and mice (R54 H, V1214E) are indicated.
Subcellular distribution of R49C-CRYAA
Site-specific mutagenesis of an expression construct encoding wild-type CRYAA46 was used to generate the R49C mutant construct. Following transfection of these constructs into HLE B-3 cells, which constitutively express alphaB-crystallin (CRYAB) but not CRYAA,47 clonal transfectants that were expressing either R49C or wild-type CRYAA at levels ranging between 0.5 and 2.0 ng/μg soluble protein, as determined by immunoblot analysis (Figure 3a), were selected for further studies. Immunoblotting further indicated that the R49C mutant, but not wild-type CRYAA, was present in the insoluble membrane fraction (Figure 3b).

Immunoblot analysis of HLE B-3 cells transfected with expression constructs encoding either wild-type or mutant CRYAA. (a) Detergent-soluble fractions showing cells transfected with vector only (Mock), a representative clonal cell line expressing wild-type CRYAA (WT) at a level of 0.5 ng/μg cellular protein and a representative clonal cell line expressing R49C-CRYAA (R49C) at a level of 0.5 ng/μg cellular protein. (b) Detergent-insoluble fractions showing that mutant CRYAA (R49C), but not wild-type CRYAA (WT) was partially insoluble.
The subcellular distribution of R49C versus wild-type CRYAA in HLE B-3 transfectants was compared using immunofluorescence confocal microscopy. Immunofluorescent labeling of wild-type CRYAA was concentrated mainly in the cytoplasm surrounding the cell nucleus (Figure 4a). Similarly, R49C-CRYAA immunofluorescence was also present in the perinuclear cytoplasm; however, 70–80% of the R49C transfectants displayed a speckled nuclear immunofluorescence (Figure 4b) that was not observed in wild-type transfectants.

Immunofluorescence confocal microscopy of HLE B-3 cells transfected with expression constructs encoding either wild-type CRYAA or the R49C mutant. (a) Localization of wild-type CRYAA (red) to the cytoplasm. (b) Localization of the R49C mutant (red) in both the cytoplasm and nucleus. Fluorescein–phalloidin staining of the f-actin cytoskeleton is shown in green. (c) Colocalization (yellow) of wild-type CRYAA (red) and endogenous CRYAB (green) in the cytoplasm. (d), Colocalization of R49C-CRYAA (red) and endogenous CRYAB (green) in both the cytoplasm and nucleus.
The distribution of endogenous CRYAB was also examined in transfected HLE B-3 cells expressing either wild-type or R49C-CRYAA. Dual immunofluorescence labeling indicated that CRYAB was colocalized with CRYAA in the perinuclear cytoplasm of wild-type transfectants (Figure 4c). In R49C transfectants, however, CRYAB immunofluorescence was both cytoplasmic and nuclear (Figure 4d), resembling that of the R49C mutant alone (Figure 4b) and suggesting that the R49C mutant may form heteromeric complexes with endogenous CRYAB.
To further examine the subcellular localization of wild-type and R49C CRYAA at the ultrastructural level, we used cryoimmunoelectron microscopy. Figure 5a shows that the immunogold labeling of wild-type CRYAA was localized to the cytoplasm of HLE B-3 transfectants. In contrast, the immunogold labeling of R49C-CRYAA was cytoplasmic and nuclear (Figure 5b), consistent with the immunofluorescent labeling (Figure 4b).

Cryoimmunoelectron microscopy of HLE B-3 cells expressing either wild-type CRYAA or the R49C mutant. (a) Immunogold labeling showed that wild-type CRYAA was restricted to the cytoplasm. (b) In contrast, R49C-CRYAA (arrows) was distributed in both the cytoplasm and the nucleus. N, nucleus. Bar, 1.5 μm, Magnification, × 50 000.
Cell death associated with R49C-CRYAA
The expression of wild-type CRYAA in HLE B-3 cells has been shown to prevent apoptosis by various inducing agents including the protein kinase inhibitor staurosporine.46,47 To compare the antiapoptotic activity of R49C-CRYAA with that of wild-type CRYAA, we measured the levels of cell death in HLE-B3 transfectants before and after staurosporine treatment. To detect apoptosis, cells were incubated with annexin V (Ax), an inner plasma-membrane phospholipid binding protein, and PI, a chromatin intercalating dye, and the percentage of labeled cells was determined by fluorescence-activated cell sorting (FACS) analysis. Basal levels of cells that were in the early stages of apoptosis (Ax-positive/PI-negative) ranged from ∼5% in mock (vector only) transfectants, <2% in wild-type CRYAA transfectants (Figure 6a and e) to ∼33% in R49C-CRYAA transfectants (Figure 6b and e). Following induction of apoptosis with staurosporine, the levels of Ax-positive/PI-negative cells increased around 10-fold to ∼50% in mock transfectants, around four-fold to ∼8% in wild-type CRYAA transfectants (Figure 6c and e) and around two-fold to ∼67% in R49C-CRYAA transfectants (Figure 6d and 6e). These observations indicated that R49C-CRYAA was not only less protective against cell death than its wild-type counterpart, but also suggested that the R49C mutant was cytotoxic.

FACS analysis of apoptosis in HLE B-3 transfectants expressing either wild-type or mutant CRYAA. Apoptosis was induced by staurosporine (STP) treatment and cells were labeled with annexin V (Ax) and propidium iodide (PI). (a) Noninduced cells (STP−) expressing wild-type CRYAA (WT). Live cells (Ax-negative/PI-negative) are shown in the bottom left quadrant. Cells in early stages of apoptosis (Ax-positive/PI-negative) are shown in the bottom right quadrant, whereas cells that have already died by apoptosis (Ax-positive/PI-positive) are shown in the top right quadrant. Necrotic cells (Ax-negative/PI-positive) are shown in the top left quadrant. (b) Noninduced cells expressing mutant CRYAA (R49C). (c) Staurosporine-induced cells (STP+) expressing WT-CRYAA. (d) Staurosporine-induced cells expressing R49C-CRYAA. (e) Relative percentages of apoptotic cells in wild-type (WT), mutant (R49C) and mock (vector only) transfectants before (STP−) and after (STP+) staurosporine induction.
Discussion
The gene for CRYAA (CRYAA) is a member of the sHSP family of molecular chaperones and encodes ∼20% of the soluble protein present in the newborn human lens,48 accounting for a substantial portion of its refractive power.40 Previously, missense (R116C) and nonsense (W9X) mutations in CRYAA have been linked with autosomal dominant15 and recessive16 forms of nonsyndromic cataract, respectively. Similarly, dominant (V124E) and recessive (R54 H) missense mutations in the murine gene for CRYAA (Cryaa) have been associated with cataract in the Aey749 and the lop1850 mouse mutants, respectively. So far, each of the dominant mutations in CRYAA (R116C) and Cryaa (V124E) lie within the phylogenetically conserved ‘alpha-crystallin domain’ of sHSPs, whereas, each of the recessive mutations in CRYAA (W9X) and Cryaa (R54 H) lie in the N-terminal region. In the present study, we have identified the first dominant mutation (R49C) in CRYAA linked with cataract, which lies outside the alpha-crystallin/sHSP core domain.
Clinical descriptions of hereditary cataract are based on the physical location, size and appearance of opacities in different developmental regions of the juvenile lens.51 The dominant R49C mutation in CRYAA described here was associated with nuclear opacities affecting the central core of the lens. Similarly, lens opacities associated with the dominant R116C mutation in CRYAA were described as zonular central nuclear15 affecting the developing core, or nucleus, and perinuclear cortex of the lens. Moreover, in the Aey749 and lop1850 mice, dominant (V124E) and recessive (R54 H) missense mutations in Cryaa, respectively, were associated with a progressive nuclear and zonular cataract. No clinical description of the autosomal recessive cataract morphology associated with the W9X mutation in CRYAA16 was published, however, the resulting premature chain termination is likely to mimic a ‘knockout’ of the protein in the homozygous state. Significantly, mice that are null for Cryaa also develop a progressive nuclear cataract.52
Transfection studies of the R49C-CRYAA mutant in HLE B-3 cells revealed a significant increase in basal (∼15-fold) and staurosporine-induced (∼8-fold) levels of cell death compared to wild-type CRYAA. Similarly, increased levels of basal (5–10-fold) and UVA-induced (∼19-fold) cell death were associated with the R116C-CRYAA mutant in HLE B-3 cells.53 In addition, bacterial expression studies of recombinant CRYAA have shown that the R116C missense substitution resulted in reduced prevention of heat-induced protein aggregation in vitro (∼four-fold) compared to wild type.54,55,56 However, this loss of chaperone-like function was largely regained when R116C-CRYAA was mixed with wild-type CRYAA and CRYAB subunits in the stoichiometric amounts (1.5:1.5:1, respectively) predicted to occur in the lens of an affected (heterozygous) individual,56 indicating the absence of dominant negative effects. Moreover, heterozygous loss of Cryaa in mice52 and, theoretically, CRYAA in humans16 is not sufficient to cause cataract. Although we cannot exclude dominant negative effects, these observations suggest that loss of chaperone-like function per se is unlikely to be the predominant mechanism underlying cataract development linked with the R49C missense mutation in CRYAA.
The high basal and induced levels of cell death associated with R49C-CRYAA compared with those of R116C-CRYAA53 suggested that the former mutant was more cytotoxic than the latter. A likely prerequisite for such toxicity is the formation of abnormal protein aggregates, which has been proposed to represent a common pathologic mechanism for protein misfolding diseases.57 Biochemical studies have shown that the aggregate size of R116C-CRYAA was ∼four-fold higher (>2 MDa) that that of wild-type CRYAA homocomplexes53 and these mutant aggregates also exhibited a significant increase (∼10-fold) in binding to cell membranes in vitro compared with wild-type CRYAA.56 Although our attempts to isolate R49C-CRYAA from HLE B-3 transfectants were unsuccessful, due to the high level of cell death, two pieces of immunochemical evidence suggest that the R49C mutant had gained more deleterious aggregation and/or binding properties. First, the R49C mutant was clearly detected in the insoluble membrane fraction obtained from HLE B-3 transfectants (Figure 3), whereas, the R116C mutant was not53 (UPA unpublished data). One factor that may contribute to insolubility of the R49C mutant is the introduction of a cysteine residue near the N-terminus. Based on the predicted topology of CRYAA, the N-terminal region appears to be involved in quaternary subunit interactions,58 raising the possibility of inappropriate disulfide bridge formation by the R49C mutant. In contrast, the R116C substitution lies within one of the seven beta-strands that structurally define the alpha-crystallin/sHSP domain58 and does not appear to generate inter-subunit disulphide bridges during aggregate formation.56 Second, when expressed in HLE B-3 cells the R49C mutant was localized to both nuclear and cytoplasmic compartments (Figure 4 and Figure 5). In contrast, the R116C-CRYAA mutant was mainly restricted to the cytoplasm.53 Interestingly, accumulation of intranuclear inclusions has long been regarded as a signature of aggregate formation in glutamine-repeat neurodegenerative disorders59 and, recently, nuclear inclusions have been associated with hereditary cataract in certain strains of mice.60 While the true pathological significance of nuclear inclusions remains controversial,59 their association with the R49C-CRYAA mutant described here suggests that certain hereditary forms of cataract and neurodegeneration in humans may be triggered by common cytotoxic mechanisms.
References
Rahi JS, Dezateaux C : The British Congenital Cataract Interest Group: Measuring and interpreting the incidence of congenital ocular anomalies: lessons from a national study of congenital cataract in the UK. Invest Ophthalmol Vis Sci 2001; 42: 1444–1448.
Sommer A, Tielsch JM, Katz J et al: Racial differences in the cause-specific prevalence of blindness in east Baltimore. N Engl J Med 1991; 325: 1412–1417.
Francois J : Genetics of cataract. Ophthalmologica 1982; 184: 61–71.
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim.
Eiberg H, Lund AM, Warburg M, Rosenberg T : Assignment of congenital cataract Volkmann type (CCV) to chromosome 1p36. Hum Genet 1995; 96: 33–38.
Ionides ACW, Berry V, Mackay DS, Moore AT, Bhattacharya SS, Shiels A : A locus for autosomal dominant posterior polar cataract on chromosome 1p. Hum Mol Genet 1997; 6: 47–51.
Khaliq S, Hameed A, Ismail M, Anwar K, Mehdi SQ : A novel locus for autosomal dominant nuclear cataract mapped to chromosome 2p12 in a Pakistani family. Invest Ophthalmol Vis Sci 2002; 43: 2083–2087.
Vanita, Singh JR, Sarhadi VK et al: A novel form of ‘central pouchlike’ cataract, with sutural opacities, maps to chromosome 15q21-22. Am J Hum Genet 2001; 68: 509–514.
Berry V, Ionides ACW, Moore AT, Plant C, Bhattacharya SS, Shiels A : A locus for autosomal dominant anterior polar cataract on chromosome 17p. Hum Mol Genet 1996; 5: 415–419.
Armitage MM, Kivlin JD, Ferrell RE : A progressively early onset cataract gene maps to human chromosome 17q24. Nat Genet 1995; 9: 37–40.
Yamada K, Tomita H, Yoshiura K et al: An autosomal dominant posterior polar cataract locus maps to human chromosome 20p12-q12. Eur J Hum Genet 2000; 8: 553–559.
Pras E, Bakhan T, Levy-Nissenbaum E et al: A gene causing autosomal recessive cataract maps to the short arm of chromosome 3. Isr Med Assoc J 2001; 3: 559–562.
Heon E, Paterson AD, Fraser M et al: A progressive autosomal recessive cataract locus maps to chromosome 9q13-q22. Am J Hum Genet 2001; 68: 772–777.
Berry V, Francis PJ, Reddy MA et al: Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet 2001; 69: 1141–1145.
Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG : Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet 1998; 7: 471–474.
Pras E, Frydman M, Levy-Nissenbaum E et al: A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci 2000; 41: 3551–3555.
Kannabiran C, Rogan PK, Olmos L et al: Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis 1998; 4: 21 (〈http://www.molvis.org/molvis/v4/p21〉).
Bateman JB, Geyer DD, Flodman P et al: A new betaA1-crystallin splice junction mutation in autosomal dominant cataract. Invest Ophthalmol Vis Sci 2000; 41: 3278–3285.
Litt M, Carrero-Valenzuela R, LaMorticella DM et al: Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet 1997; 6: 665–668.
Gill D, Klose R, Munier FL et al: Genetic heterogeneity of the Coppock-like cataract: a mutation in CRYBB2 on chromosome 22q11.2. Invest Ophthalmol Vis Sci 2000; 41: 159–165.
Vanita, Sarhadi V, Reis A et al: A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J Med Genet 2001; 38: 392–396.
Mackay DS, Boskovska OB, Knopf HLS, Lampi KJ, Shiels A : A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet 2002; 71: 1216–1221.
Heon E, Priston M, Schorderet DF et al: The gamma crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet 1999; 65: 1261–1267.
Stephan DA, Gillanders E, Vanderveen D et al: Progressive juvenile-onset punctate cataracts caused by mutation of the gammaD-crystallin gene. Proc Natl Acad Sci USA 1999; 96: 1008–1012.
Kmoch S, Brynda J, Asfaw B et al: Link between a novel human gammaD-crystallin allele and a unique cataract phenotype explained by protein crystallography. Hum Mol Genet 2000; 9: 1779–1786.
Ren Z, Li A, Shastry BS et al: A 5-base insertion in the gammaC-crystallin gene is associated with autosomal dominant variable zonular pulverulent cataract. Hum Genet 2000; 106: 531–537.
Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S : A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant ‘zonular pulverulent’ cataract on chromosome 1q. Am J Hum Genet 1998; 62: 526–532.
Berry V, Mackay D, Khaliq S et al: Connexin50 mutation in a family with congenital ‘zonular nuclear’ pulverulent cataract of Pakistani origin. Hum Genet 1999; 105: 168–170.
Polyakov AV, Shagina IA, Khiebnikova OV, Evgrafov OV : Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract. Clin Genet 2001; 60: 476–478.
Mackay D, Ionides A, Kibar Z, Rouleau G, Shiels A, Bhattacharya S : Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 1999; 64: 1357–1364.
Rees MI, Watts P, Fenton I et al: Further evidence of autosomal dominant congenital zonular pulverulent cataracts linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3). Hum Genet 2000; 106: 206–209.
Francis P, Chung JJ, Yasui M et al: Functional impairment of lens aquaporin in two families with dominantly inherited cataracts. Hum Mol Genet 2000; 9: 2329–2334.
Pras E, Levy-Nissenbaum E, Bakhan T et al: A missense mutation in the LIM2 gene is associated with autosomal recessive presenile cataract in an inbred Iraqi Jewish family. Am J Hum Genet 2002; 70: 1363–1367.
Conley YP, Erturk D, Keverline A et al: A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet 2000; 66: 1426–1431.
Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M : Autosomal dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet 2000; 66: 1432–1436.
Semina EV, Ferrell RE, Mintz-Hittner HA et al: A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet 1998; 19: 167–170.
Bu L, Jin Y, Shi Y et al: Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 2002; 31: 276–278.
Okano Y, Asada M, Fujimoto A et al: A genetic factor for age-related cataract: identification and characterization of a novel galactokinase variant, ‘Osaka,’ in Asians. Am J Hum Genet 2001; 68: 1036–1042.
Brooks DG, Manova-Todorova K, Farmer J et al: Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome. Invest Ophthalmol Vis Sci 2002; 43: 1121–1126.
Horwitz J : The function of alpha-crystallin in vision. Semin Cell Dev Biol 2000; 11: 53–60.
Dib C, Faure S, Fizames C et al: A comprehensive genetic map the human genome based on 5264 microsatellites. Nature 1996; 380: 152–154.
Lathrop GM, Lalouel JM, Julier C, Ott J : Strategies for multipoint linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
Andley UP, Mathur S, Griest TA, Petrash JM : Cloning, expression, and chaperone-like activity of human alphaA-crystallin. J Biol Chem 1996; 271: 31973–31980.
Jaworski CJ, Piatigorsky J : A pseudo-exon in the functional human alpha A-crystallin gene. Nature 1989; 337: 752–754.
Collins JS, Schwartz CE : Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet 2002; 71: 1251–1252.
Andley UP, Song Z, Wawrousek EF, Bassnett S : The molecular chaperone alphaA-crystallin enhances lens epithelial cell growth and resistance to UVA stress. J Biol Chem 1998; 273: 31252–31261.
Andley UP, Song Z, Wawrousek EF, Fleming TP, Bassnett S : Differential protective activity of alphaA- and alphaB-crystallin in lens epithelial cells. J Biol Chem 2000; 275: 36823–36831.
Lampi KJ, Ma Z, Shin M et al: Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes the identification of the major proteins in young human lens. J Biol Chem 1997; 272: 2268–2275.
Graw J, Loster J, Soewarto D et al: Characterization of a new, dominant V124E mutation in the mouse alphaA-crystallin-encoding gene. Invest Ophthalmol Vis Sci 2001; 42: 2909–29015.
Chang B, Hawes NL, Roderick TH et al: Identification of a missense mutation in the alphaA-crystallin gene of the lop18 mouse. Mol Vis 1999; 5: 21 (〈www.molvis.org/molvis/v5/p21〉).
Phelps-Brown N, Bron AJ : Lens disorders: a clinical manual of cataract diagnosis. Oxford, UK: Butterworth-Heineman Ltd., 1996.
Brady JP, Garland D, Douglas-Tabor Y, Robison WG, Groome A, Wawrousek EF : Targeted disruption of the mouse alpha A-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein alpha B-crystallin. Proc Natl Acad Sci USA 1997; 94: 884–889.
Andley UP, Patel HC, Xi JH : The R116C mutation in alpha A-crystallin diminishes its protective ability against stress-induced lens epithelial cell apoptosis. J Biol Chem 2002; 277: 10178–10186.
Kumar LVS, Ramakrishnan T, Rao CM : Structural and functional consequences of the mutation of a conserved arginine residue in alphaA and alphaB crystallins. J Biol Chem 1999; 274: 24137–24141.
Shroff NP, Cherian-Shaw M, Bera S, Abraham EC : Mutation of R116C results in highly oligomerized alpha A-crystallin with modified structure and defective chaperone-like function. Biochemistry 2000; 39: 1420–1426.
Cobb BA, Petrash JM : Structural and functional changes in the alphaA-crystallin R116C mutant in hereditary cataracts. Biochemistry 2000; 39: 15791–15798.
Bucciantini M, Giannoni E, Chiti F et al: Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002; 416: 507–511.
Koteiche HA, Mchaourab HS : Folding pattern of the alpha-crystallin domain in alphaA-crystallin determined by site-directed spin labeling. J Mol Biol 1999; 294: 561–577.
Ross CA : Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 2002; 35: 819–822.
Sandilands A, Hutcheson AM, Long HA et al: Altered aggregation properties of mutant gamma-crystallins cause inherited cataract. EMBO J 2022; 21: 6005–6014.
Acknowledgements
We thank the family for participating in this study, Dr Olivera Boskovska for help with ascertaining the family, William Walker, Jing-Hua Xi and Harendra Patel for excellent technical assistance, Dr Wandy Beatty for cryoimmunoelectron microscopic imaging, Cheryl Shomo for help with graphics and an anonymous reviewer for constructive comments. This work is supported by NIH/NEI Grants EY12284 (AS), EY05681 (UPA), EY02687 and Research to Prevent Blindness.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mackay, D., Andley, U. & Shiels, A. Cell death triggered by a novel mutation in the alphaA-crystallin gene underlies autosomal dominant cataract linked to chromosome 21q. Eur J Hum Genet 11, 784–793 (2003). https://doi.org/10.1038/sj.ejhg.5201046
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201046
Keywords
This article is cited by
-
Placing human gene families into their evolutionary context
Human Genomics (2022)
-
BMP3 is a novel locus involved in the causality of ocular coloboma
Human Genetics (2022)
-
Changes in relative histone abundance and heterochromatin in αA-crystallin and αB-crystallin knock-in mutant mouse lenses
BMC Research Notes (2020)
-
The genetic landscape of crystallins in congenital cataract
Orphanet Journal of Rare Diseases (2020)
-
Identification and expression analysis of multiple small heat shock protein genes in spruce budworm, Choristoneura fumiferana (L.)
Cell Stress and Chaperones (2018)
