
Abstract
Mutations in the WT1 gene causing Wilms tumors were first reported in WAGR syndrome (Wilms tumor, Aniridia, Genitourinary malformation, mental Retardation) and Denys Drash syndrome (pseudohermaphroditism, Wilms tumor, nephropathy), but only in a few patients with hypospadias and cryptorchidism without other signs of Denys Drash (DDS) or WAGR syndrome WT1 mutations were identified. We report a boy, who was born in 1989 with hypospadias and bilateral cryptorchidism. Previous karyotyping and endocrine studies had ruled out any known cause of male pseudohermaphroditism. Subsequently, he developed a bilateral Wilms tumor, which was detected by palpation at the age of 15 months during a routine visit by the general pediatrician. Because of its extensive size, surgery and chemotherapy were needed for treatment. Analysis of the WT1 gene was performed 5 y after diagnosis and revealed a C to T transition in one allele generating a stop codon at codon 362 and subsequently leading to a truncated protein with loss of its ability to bind to DNA. No signs of DDS or WAGR syndrome are present in the boy. The work up of this patient and the so far known few comparable cases from the literature lead to the conclusion that in newborns with severe urogenital malformations not due to known chromosomal or endocrine disorders mutational screening of the WT1 gene should be performed, to evaluate the high risk of developing a Wilms tumor. We favor mutational screening in these patients as an easy tool for investigation, because in the future it will probably decrease the necessity of frequent control visits in patients without a WT1 mutation.
Similar content being viewed by others

Main
The Wilms tumor suppressor gene 1 (WT1) encodes a transcription factor playing a major role during early embryonal development of the kidneys and the urogenital system in mammals. It is highly expressed in the early developing kidney and the gonads (1,2). WT1 is located on chromosome 11p13, consists of 10 exons, covers 50 kb and belongs to the zinc finger family of transcription factors (3,4). It was detected by positional cloning from the genomic DNA of patients with WAGR syndrome (Wilms tumor, Aniridia, Genitourinary malformations, mental Retardation) carrying deletions of the 11p13 region. Since then, apart from deletions of the WT1 gene in WAGR syndrome and tumor-specific alterations in Wilms tumors, germline mutations of the WT1 gene were detected causing early Wilms tumors in patients with Denys Drash syndrome (DDS), another syndrome with inborn urogenital malformations consisting of male pseudohermaphroditism, nephropathy and Wilms tumor (5,6). Until now almost 150 cases of DDS have been described, and mutations of the WT1 gene have been detected in 48 to date genetically analyzed cases (7–9). In three analyzed cases no mutations in the coding sequence of the WT1 gene were found (10,11).
Additionally, germline mutations of WT1 were found so far only in six children, with severe urogenital malformations and no other signs of DDS or WAGR syndrome, who developed Wilms tumor at an early age (12–15). This study reports the diagnosis in a boy with severe hypospadias and bilateral cryptorchidism, who was born 1 year before cloning of the WT1 gene and developed a bilateral large Wilms tumor at the age of 15 months, which required extensive surgery and chemotherapy for treatment.
THE PATIENT
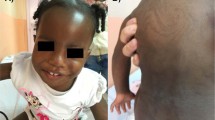
The patient was born in 1989 as the second child after an uneventful pregnancy (1 y before the cloning of the WT1 gene). He presented at birth with severe hypospadias and bilateral cryptorchidism (Fig. 1). The karyotype was normal male (46, XY). Penis size was normal, the scrotum was normally rugated and no hyperpigmentation was visible. Defects of testosterone synthesis, androgen-receptor defects and congenital adrenal hyperplasia were ruled out by biochemical and molecular genetic studies. Sonography of the kidneys revealed a duplication of the left kidney and left ureter (Fig. 2). A mild vesicoureteral reflux was treated with low-dose antibiotics.

External genitalia of the patient.

Internal genitalia of the patient.
At the age of 15 months during a routine control by the general pediatrician, abdominal extension was observed and a giant abdominal mass (18 cm × 18 cm) reaching from the upper left to the middle abdomen was palpated. Subsequently bilateral Wilms tumor of stromal-predominant histology was diagnosed. For treatment three courses of chemotherapy (1. Actinomycin, Vincristin, 2. Cyclophosphamide, Adriamycin, Vincristin, Actinomycin, 3. VP 16, Carboplatin, Ifosfamid, Adriamycin, Vincristin) and left-sided nephrectomy was needed.
During surgery a thorough examination of the internal genitalia revealed both testes being attached to the frontal lower poles of the kidneys, and rudiments of a uterus and vagina were seen. The left testis was removed and the right testis was brought and fixed in the scrotum.
At the age of 6 and 7 y, surgical corrections of the urogenital malformation were performed. The rudimentary vagina was excised and the right endocrine nonfunctional testis, as proven by negative HCG test, was removed. Until now (7 y after therapy), no relapse of Wilms tumor and no further signs of DDS or WAGR syndrome were detectable.
METHODS AND RESULTS
Analysis of the WT1 gene was performed as previously described (12). The patient's genomic DNA was isolated from peripheral leukocytes, and subsequently PCR-SSCP analysis of all 10 exons of the WT1 gene was performed. An aberrant banding pattern was observed in exon 8. Sequencing revealed a C to T transition in one allele at position 1084 resulting in a stop codon (362Arg to Stop). During translation this mutation leads to a truncated protein missing the last three zink fingers which are necessary for DNA binding (Fig. 3) (16).

(A) Sequence analysis of the patient and a healthy control showing the heterozygous point mutation (1084 C->T) of the patient. (B) WT1 gene and protein structure including the mutation of the patient, which is leading to a truncated protein (25).
DISCUSSION
WAGR syndrome and DDS are rare syndromes with association of urogenital malformations and Wilms tumor due to deletions and mutations of the WT1 gene. WAGR syndrome has been observed in 2% of Wilms tumor cases. Hypospadias and cryptorchidism were reported in up to 4.4% in children with Wilms tumor (17–19). In a recent study in 4% of patients with urogenital anomalies and Wilms tumor, WT1 mutations were detected (20). The boy described in this study so far is one of the very rare published cases with severe urogenital malformations and early developing Wilms tumor due to a mutation in the WT1 gene without signs of DDS or WAGR syndrome.
All patients with germline mutations of the WT1 gene are only heterozygous for the mutated allele. Mice, homozygously deleted for WT1, fail to develop kidneys or gonads, verifying a critical role for WT1 in urogenital development (21). There are three major types of mutations: 1. missense mutations altering amino acids that directly interact with the DNA target, 2. substitutions of amino acids involved in zinc complexing, and 3. nonsense mutations leading to the removal of at least two zinc fingers. As shown in transfection assays, WT1 mutations can act in a dominant negative fashion with loss of normal DNA binding of WT1 through loss or conformational change of the DNA binding zinc finger region and thereby decreased transcriptional suppression activity (16,22). It has been suggested that in most cases with a heterozygous WT1 mutation, a second hit leading to loss of heterozygosity seems to be important for tumor genesis, because almost all Wilms tumors in those patients are homozygous for the mutated allele (12,23,24).
From this study and our current knowledge, we have to conclude that molecular genetic studies of the WT1 gene are mandatory in the work up of newborns with severe urogenital anomalies. As children with severe urogenital malformations due to a heterozygous mutation in the WT1 gene are at high risk for the development of a Wilms tumor at an early age, efforts should be taken to identify those children immediately after birth to follow them closely. In children with severe hypospadias and cryptorchidism after exclusion of other chromosomal, endocrine, or androgen-receptor-mediated disorders, either ultrasound controls of the kidneys in brief intervals (4 weekly) or molecular analysis of the WT1 gene must be performed.
As mutational screening of the WT1 gene by means of PCR-SSCP analysis or direct sequencing have become easy tools of investigation, we favor these methods to identify children being at high risk for development of a Wilms tumor. This will result in an earlier diagnosis and treatment of this malignant disease. Moreover in the future frequent ultrasound controls of the kidneys probably can be limited to individuals with mutations of the WT1 gene, in the remaining patients without WT1 mutations ultrasound monitoring may be reduced to longer (e.g. 3-6 months) intervals. This approach has the advantage of a better cost effectiveness by reduction of the otherwise necessary frequent control visits in a large number of patients.
Abbreviations
- WT1:
-
Wilms tumor suppressor gene 1
- DNA:
-
desoxyribonucleid acid
- kb:
-
kilobases
- WAGR:
-
Wilms tumor aniridia genitourinary malformation mental retardation
- DDS:
-
Denys Drash syndrome
- HCG:
-
human chorionic gonadotropin
- PCR:
-
polymerase chain reaction
- SSCP:
-
single stranded conformational polymorphism
References
Armstrong JF, Pritchard-Jones K, Bickmore WA, Hastie ND, Bard JBL 1992 The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev 40: 85–97
Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D, van Heyningen V, Hastie N 1990 The candidate Wilms' tumour gene is involved in genitourinary development. Nature 346: 194–197
Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE 1990 Isolation and characterisation of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell 60: 509–520
Gessler M, Poustka A, Cavenne W, Neve RL, Orkin SH, Bruns GAP 1990 Homozygous deletion in Wilms tumors of a zinc-finger gene identified by chromosome jumping. Nature 343: 774–778
Drash A, Sherman F, Hartmann WH, Blizzard RM 1970 A syndrome of pseudohermaphroditism, Wilms tumour, hypertension and degenerative renal disease. J Pediatr 76: 585–593
Denys P, Malvaux P, van den Berghe H, Tanghe W, Proesmans W 1967 Association d'un syndrome anatomo-pathologique de pseudohermaphroditisme masculin, d'une tumeur de Wilms, d'une nephropathie parenchymateuse et d'un mosaicisme XX/XY. Arc Fr Pediatr 24: 729–39
Mueller RF 1994 The Denys Drash syndrome J Med Genet 31: 471–477
Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton C Junien, Habib R, Fouser L, Fine RN, Silverman BL, Haber DA, Housman D 1991 Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash Syndrome. Cell 67: 437–447
Little M, Wells C 1997 A clinical overview of WT1 gene mutations. Hum Mutat 9: 209–225
Nordenskjold A, Friedman E, Anvret M 1994 WT1 mutations in patients with Denys-Drash syndrome - a novel mutation in exon 8 and paternal allele origin. Hum Genet 93: 115–120
Baird PN, Santos A, Groves N, Jadresic L, Cowell JK 1992 Constitutional mutations in the WT1 gene in patients with Denys-Drash syndrome. Hum Mol Genet 1: 301–305
Schumacher V, Schneider S, Figge A, Wildhardt G, Harms D, Schmidt D, Weirich A, Ludwig R, Royer-Pokora B 1997 Correlation of germ-line mutations and two-hit inactivation of the WT1 gene with Wilms tumors of stromal-predominant histology. Proc Natl Acad Sci 94: 3972–3977
Huff V, Jaffe N, Saunders GF, Strong LC, Villalba F, Ruteshouser EC 1995 WT1 exon1 deletion/insertion mutations in Wilms' tumor patients associated with di- and trinucleotide repeats and deletion hotspot consensus sequences. Am J Hum Genet 56: 84–90
Nordenskjöld A, Friedman E, Sandstedt B, Söderhall S, Anvret M 1995 Constitutional and somatic mutations in the WT1 gene in Wilms' tumor patients. Int J Cancer 63: 516–522
Pelletier J, Bruenig W, Li FP, Haber DA, Glaser T, Housman DE 1991 WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumor. Nature 353: 431–434
Little M, Holmes G, Bickmore W, van Heyningen V, Hastie N, Wainwright B 1995 DNA binding capacity of the WT1 protein is abolished by Denys-Drash syndrome WT1 point mutations. Hum Mol Genet 3: 351–358
Matsunaga E 1981 Genetics of Wilms' tumor. Hum Genet 57: 231–246
Miller RW, Fraumeni JF Jr, Manning MD 1964 Association of Wilms' tumor with aniridia, hemihypertrophy and other congenital malformations. N Engl J Med 270: 922–927
Pendergrass TW 1976 Congenital anomalies in children with Wilms' tumor: a new survey. Cancer 1: 403–408
Li FP, Breslow NE, Morgan JM, Ghahremani M, Miller GA, Grundy PE, Green DM, Diller LR, Pelletier J 1996 Germline WT1 mutations in Wilms' tumor patient: preliminary results. Med Pediatr Oncol 27: 404–407
Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R 1993 WT-1 is required for early kidney development. Cell 74: 679–691
Reddy JC, Morris JC, Wang J, English MA, Haber DA, Shi Y, Licht JD 1995 WT1-mediated transcriptional activation is inhibited by dominant negative mutant protein. J Biol Chem 270: 10878–10884
Knudson AG Jr, Strong LC 1972 Mutation and cancer: a model for Wilms' tumor of the kidney. J Natl Cancer Inst 48: 313–324
Knudson AG Jr 1971 Mutation and cancer: statistical study. Proc Natl Acad Sci USA 68: 820–823
Bruening W, Winnett E, Pelletier J 1995 Wilms' tumour: a paradigm for insights into development and cancer. Cancer Inv 13: 431–443
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Köhler, B., Schumacher, V., Schulte-Overberg, U. et al. Bilateral Wilms Tumor in a Boy with Severe Hypospadias and Cryptorchidism Due to a Heterozygous Mutation in the WT1 Gene. Pediatr Res 45, 187–190 (1999). https://doi.org/10.1203/00006450-199902000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199902000-00005
