
Abstract
The genetic differentiation and the phylogenetic relationships of eight Atherina boyeri Greek populations have been investigated at the mtDNA level. The populations studied are from two different lakes, a lagoon, the interface zone between the lagoon and the sea, and four marine sites. RFLP analysis of three mtDNA segments (12s rRNA, 16s rRNA and D-loop) amplified by PCR was used. Six, seven and eight restriction enzymes were found to have at least one recognition site at 12s rRNA, 16s rRNA and D-loop respectively. Twenty-one different haplotypes were detected among the populations studied. Several restriction patterns were revealed. These patterns can be used for the discrimination of the populations living in the sea (‘marine’ type populations) from the others inhabiting the lagoon and the lakes (‘lagoon’ type populations). The estimated net nucleotide sequence divergence between the populations examined ranged from 0 to 10.385%, while the Nst value of 0.92 indicates the existence of high interpopulation genetic differentiation. This high degree of differentiation detected between the ‘lagoon’ and ‘marine’ type populations makes the classification of these two types of populations as a single taxon questionable.
Similar content being viewed by others

Introduction
The Atherinidae family is represented by a single genus in Europe consisting of three species, Atherina boyeri Risso 1810, Atherina hepsetus L.1758 and Atherina presbyter Cuvier 1829 (Kiener and Spillman, 1969). Atherina boyeri is commonly found in the coastal waters of the east-Atlantic Ocean as well as in the Mediterranean, Black, Caspian and Aral seas (Gon and Ben-Tuvia, 1983).
Atherina boyeri is an extremely euryhaline teleost fish which inhabits coastal and estuarine waters as well as lagoon shallow brackish water ecosystems and inland waters. This small fish forms local semi-isolated populations which may be different with respect to their population biology and morphology (Henderson and Bamber, 1987). In spite of the general morphological similarities exhibited by A. boyeri populations, studies based on morphometric and meristic characters of marine and lagoon populations revealed the existence of two different ecotypes (Kartas and Trabelsi, 1990; Focant et al, 1992). Moreover local marine populations, whose morphology distinguishes them clearly from their neighbours, have also been observed by Trabelsi et al (1994).
High degree of genetic differentiation has been revealed, through protein electrophoresis, among A. boyeri populations (Berrebi and Britton-Davidian, 1980; Creech, 1991). Moreover, Focant et al (1999), who studied the electrophoretic patterns of some small muscle proteins (paralvoumines), found differences between populations living in the Rhone delta (lagoon) and a marine population living nearby.
The differentiation revealed by morphological and electrophoretic studies has led to a confusion in the taxonomic status of this species. Historically, a number of species and subspecies have been recognized, which are now included within the species A. boyeri (Kiener and Spillman, 1969; Bamber and Henderson, 1985). For this reason A. boyeri is viewed by many investigators as a polymorphic complex (Focant et al, 1999).
On the other hand, molecular biology has provided new and more powerful tools for population genetic studies. This is because DNA analysis results in a better resolution of genetic variation than does allozyme electrophoresis. MtDNA represents a significant marker system for use in population and phylogenetic studies. MtDNA is generally maternally inherited without genetic recombination. The evolutionary rate as well as the genetic differentiation of mtDNA among populations is thought to be approximately 5–10 times higher than that exhibited by nuclear genes (Birky et al, 1983, 1989; Wilson et al, 1985). An extensive review of the advantages of mtDNA as a tool for population genetic analysis has been provided by Avise (1991, 1994).
Despite the advantages of mtDNA analysis in population genetic studies, no such studies concerning A. boyeri populations have been presented so far in the literature. For this reason we investigate the genetic structure of A. boyeri populations using restriction fragment length polymorphism (RFLP) analysis of mtDNA. The Greek populations used in the present study originated from Messologi lagoon, from the freshwater lakes of Trichonida and Kaiafas and from some marine sites of the western region of Greece. With this experimental system, we attempt to answer the following questions:
-
1
Are there any important genetic differences between the populations living in the sea and those inhabiting the lagoon and the lakes?
-
2
Are there any important genetic differences between the distant lake populations of Trichonida and Kaiafas.
-
3
Is the A. boyeri population in Messologi lagoon a single homogeneous one or is it a mixed population consisting of specimens originating from both the lagoon and the sea?
-
4
Is the population living in the lagoon genetically similar to that living in the nearby lake of Trichonida since the lagoon and the lake are more or less connected with each other as well as with the sea?
-
5
Is there a single homogeneous population living in the sea or are there local, genetically differentiated, marine populations?
Materials and methods
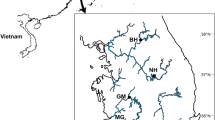
Samples from eight different populations of A. boyeri were collected from the western region of Greece (Figure 1) using traps. A total number of, 196 individuals were sampled from the lakes of Trichonida (TRI) in the Central Greece and Kaiafas (KAI) in Peloponnese, from Messologi lagoon (MES), from the interface zone between the lagoon and the sea (MET), as well as from four different marine sites; one from the coast of central Greece, Monastiraki (MON) and three from the Peloponnese coast, namely Kiparissia (KIP), Kalamaki (KAL) and Diminio (DIM).

Sampling sites: TRI, Lake Trichonida; KAI, Lake Kaiafas; MES, Messolongi Lagoon; MET, Population from the interface zone between the lagoon and the sea; MON, Monastiraki; KAL, Kalamaki; DIM, Diminio; KIP, Kiparisia.
All individuals were transferred in dry ice to the laboratory and stored at −80°C until used. Total DNA was isolated from each individual according to the protocol of Jackson et al (1991), after minor modifications. About 50 mg of muscle tissue was ground in 400 μl homogenizing buffer (20 mM Tris-HCl pH: 7.5, 200 mM NaCl, 20 mM EDTA). Then 100 μl of 10% SDS and 5 μl proteinase K (20 mg/ml) were added and the mixture was incubated at 50°C for 30 min. DNA was purified with standard phenol: chloroform extractions, precipitated with ice cold absolute ethanol and resuspended in 50 μl TE (Tris-EDTA pH: 8) buffer.
For the amplification of the three mtDNA segments studied, three sets of primers were used. MtDNA variation was then analyzed by RFLPs, performed on PCR-amplified products. The primers used for the 12s rRNA and 16s rRNA mtDNA segments were the universal primers 12SAL-12SBH and 16SARL-16SBRH described by Palumbi et al (1991), while the primers for the D-loop region were those described by Lee et al (1995). Double-stranded DNA amplifications were performed in 50 μl volumes containing 2 units of Taq polymerase, 5 μl of 10 × reaction buffer provided by the manufacturer Promega, 0.2–0.5 mM dNTPs mix, 1.5 mM MgCl2, approximately 100–200 ng DNA and 0.2–0.5 μM of each primer. PCR amplification conditions were as follows: one preliminary denaturation step at 94°C for 4 min followed by 35 PCR cycles. Strand denaturation was at 94°C for 1 min, annealing at 57°C (12s rRNA), 50°C (16s rRNA) and 51°C (D-loop) for 1 min and primer extension at 72°C for 1.5 min. A final extension at 72°C for 5 min was performed.
Amplified mtDNA segments from three individuals of each population were digested with at least 25 restriction enzymes in order to check the presence of recognition sites. The informative restriction enzymes were then applied to approximately 20 individuals from each population except in the cases of TRI (40 specimens) and MET (35 specimens). The informative restriction enzymes used for the 12s rRNA gene segment were: BstN I, Dde I, Kpn I, Nla III, Rsa I, Taq I.; for the 16s rRNA gene segment were: BstN I, BstUI, Dde I, Hae III, Hpa II, Nla III and Taq I., and for the D-loop were: Aci I, BstN I, Dde I, Hind III, Hinf I, Hpa II, Rsa I and Taq I.
The digested segments were then separated electrophoretically on 2% agarose gels, in 1 × TBE buffer, stained with ethidium bromide and visualized under UV light. The sizes of DNA fragments were compared to the PCR marker (Promega) run on the same gel. A letter, in order of appearance identified single restriction patterns. Composite genotypes for each individual were then defined from all the restriction patterns of the three mtDNA gene segments. The restriction fragment data were converted to restriction site data (gain or loss of restriction site).
The degree of nucleotide divergence was estimated using the REAP computer package (McElroy et al, 1991). Two methods were used for the construction of the phylogenetic trees: the UPGMA method (Sneath and Sokal, 1973), based on net nucleotide divergence between the populations, and Dollo parsimony analysis (Farris, 1977), based on the presence or absence of restriction sites. The confidence of the branches was evaluated (1000 replicates) by the application of the bootstrap method (Felsenstein, 1985). All the above methods are available in the PHYLIP (version 3.4) computer package (Felsenstein, 1993). Trees were drawn using the TREEVIEW program (Page, 1996). The degree of geographical heterogeneity of mtDNA haplotype distribution was assessed using a χ2 statistic as described by Roff and Bentzen (1989). The significance level was obtained by 1000 Monte Carlo randomization using the MONTE program from the REAP package. NST (Lynch and Crease, 1990) was used to estimate the degree of population subdivision at the nucleotide level. The resulting index gives the ratio of the average genetic distance between genes from different populations relative to that among genes in the population. Values of NST range from 0 (no population subdivision) to 1 (complete population subdivision).
Results
The sizes of the PCR-amplified mtDNA segments for all populations studied, were found to be about 450 bp for 12s rRNA, 600 bp for 16s rRNA and 580 bp for D-loop. Six, seven and eight restriction enzymes had at least one recognition site in the amplified 12s rRNA, 16s rRNA and D-loop segments, respectively. The restriction enzymes used generated a total of 56 restriction sites corresponding to an estimated fragment of 240 bp analyzed. Fragment patterns produced by each restriction enzyme for the three mtDNA segments studied are shown in Tables 1, 2 and 3.
Several restriction enzymes produced population specific patterns. Thus, after the digestion of16s rRNA gene segment with the restriction enzymes BstN I, Hae III, HpaIII, NlaIII, the digestion of 12s rRNA gene segment with the restriction enzymes Kpn I, NlaIII, Rsa I and the digestion of D-loop segment with the restriction enzymes Aci I, BstN I, DdeI, Hinf I and Rsa I the restriction patterns obtained distinguish Peloponnese marine populations from the lagoon and the lakes populations. The aformentioned 12 patterns can be characterized as diagnostic (Table 4). Furthermore, nine such diagnostic patterns were also revealed between the ‘lagoon’ and the Monastiraki marine population, while only one can be used for the distinction of the Peloponnese marine populations from that of Monastiraki. Intrapopulation variation was also observed in all populations examined except those of MES, MET and KAI.
The 21 different haplotypes (composite genotypes) which were detected in the eight populations studied, and the haplotype frequencies, and the haplotype and nucleotide diversity values are presented in Table 4.
Estimated nucleotide sequence divergence among the 21 haplotypes was found to range from 0.000 to 15.890% (data not shown). The net interpopulation nucleotide divergence estimates among the eight populations studied, varied from 0.000% (between MES, MET and KAI) to 10.385% (between the populations MES, MET, KAI and KIP; Table 5).
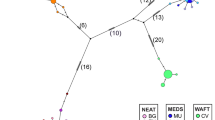
The distance matrix of net interpopulation nucleotide divergence was used to construct an UPGMA tree relating the eight populations studied (Figure 2). The populations were clustered in two distinct clades. The first one contains the populations living in the lagoon (MES), in the interface zone (MET) as well as in the lakes Trichonida (TRI) and Kaiafas (KAI); the second clade contains the four marine populations. Into this clade the three populations living in Peloponnese coast (KAL, KIP, DIM) seem to be more close and grouped together, while the marine population from Monastiraki (MON) is more distinct. Analogous situation there exist in the majority-rule consensus tree provided by Dollo parsimony method (Figure 3), where the first clade contains the most common haplotype 1 found in the four populations from the lakes, the lagoon and the interface zone, as well as haplotypes 2, 3, 4 and 5, found in the lake Trichonida. The second clade contains all the marine populations. In that clade the haplotype 6, as well as the haplotypes 9, 10, 11 found in Monastiraki, seem to be more distinct from the haplotypes found in the three populations of Peloponnese coast which are grouped together. It has to be noted that the haplotype 7, 8, and 12 originated from Monastiraki are closer to that of marine populations from Peloponnese, being their sister group.

UPGMA (Sneath and Sokal, 1973) dendrogram, based on the net nucleotide divergence, showing the relationships between the eight populations studied.

Dollo parsimony (Farris, 1977) dendrogram showing the relationships between the 21 mtDNA haplotypes detected. Numbers indicate the percentage out of 1000 bootstrap replicates that each node occurred in this majority-rule consensus tree.
Statistically significant differences in haplotype frequencies among all populations were observed (χ2 = 141.41, P < 0.001). The estimated NST value of 0.92 shows that only 8% of the overall genetic diversity observed, was within populations as opposed to 92% among populations.
Discussion
The great variability revealed by various studies based on morphological and protein electrophoretic data has led to a confusion about the taxonomic status of A. boyeri. This situation is explored by our data based on the study of mtDNA variation among eight populations of A. boyeri.
Our results revealed the existence of two clearly distinguishable types of populations. The first one consists of the populations inhabiting the lagoon, the lakes and the interface zone between the lagoon and the sea (‘lagoon’ type), while the second one contains the populations living in the sea (‘marine’ type). The assumption about the existance of ‘lagoon’ type population is strengthened by the observation that even distant lake populations as, for example, those of TRI and KAI were found to exhibit an identical composite haplotype. This fact supports the view that these ecosystem promote adaptive divergence which leads to the generation of specific ‘lagoon’ type ecotypes. An analogous situation has been described by Beheregaray and Sunnucks (2001) for Odontesthes argentinensis. It is known that the lake of Trichonida communicates, through various channels, with the lagoon and so the population existing in the lake seems to originate from the lagoon. The sand smelt from the interface zone between the lagoon and the sea (MET) are identical with those living in the lagoon and the lake. An analogous situation, where a population from the interface zone originated from the single homogenous population living in the lagoon of Camargue has been described by Focant et al (1999).
The low levels of genetic variability revealed in the ‘lagoon’ populations are consistent with the observations of Cognetti (1994) and Cognetti and Maltagliati (2000) who found that brackish populations had lower levels of genetic variability than their marine counter-parts. This situation could be attributed to the more stable and larger evolutionary population size of marine populations (DeWoody and Avise, 2000).
As regards the marine populations (KIP, KAL, DIM) from the Peloponnese coast, it must be underlined they share the same main composit haplotype. On the other hand the population originated from the coast of Central Greece (MON) is quite different from the ‘lagoon’ type, being closer to the other marine populations. This population seems to be a local one having no specimens originating either from the lagoon or the other marine populations. This situation supports the assumption according to which the observed genetic differentiation could be promoted by marine niches.
The levels of divergence observed between the 21 mtDNA haplotypes studied range from 0.000 to15.890% (data not presented) which is higher than that for the intraspecific level (10%) reported by Billington and Hebert (1991). The observed values of net interpopulation divergence revealed between the ‘lagoon’ and the three ‘marine’ populations from the coast of Peloponnese (ranging from 10.150 to 10.385) as well as the values between the ‘lagoon’ and the marine population of Monastiraki (from 8.830 to 9.041%) were high relative to other studies. Bernatchez et al (1991), suggest that above 5% divergence, the existence of undetectable homoplasy in either restriction or fragment data analysis, leads to an underestimation of the real values and thus our estimated values should be considered as minima.
The clear genetic differentiation between ‘lagoon’ and ‘marine’ populations is also highlighted by all the dendrograms presented in this study as well as by the great number of diagnostic patterns revealed between them. Moreover, the genetic differentiation between the populations studied is also evident by the NST value of 0.92 according to which 92% of the overall genetic diversity observed was among populations. The estimated NST value is in general higher than those cited by Lynch and Crease (1990) for various groups of invertebrates and vertebrates, but such high values have been reported, after RFLP analysis of mtDNA, for some freshwater fish (Apostolidis et al, 1996; Ismiridou et al, 1998; Triantaphyllidis et al, 1999). Due to the high nucleotide sequence divergence value, the estimated time of divergence between ‘marine’ and ‘lagoon’ type populations of A. boyeri is very large even if we accept the most conservative value for this estimate presented in the literature (Bernatchez et al, 1991, 2% per million years).
It is known that A. boyeri has a wide geographical range but its preferred habitat is sheltered inshore waters and coastal lagoons. This situation leads to the sub- division of this species into isolated or semi-isolated populations separated by unsuitable habitat. Many of these populations are quite small, producing ideal conditions for random genetic drift, while selection forces cannot be excluded and thus it is not surprising that the species under study displays great variability in morphological and meristic characters (Henderson and Bamber, 1987). It is also stated by the above authors that A. boyeri is best viewed as being on the brink of speciation. On the other hand, based on the continuous nature of the meristic variability, taxonomists assume that A. boyeri populations form a single species suggesting that much of the variability is phenotypic rather than genetic. It must be noted that although extensive variation in the levels of the mean genetic distance exists within and among major vertebrates groups (Jones and Avise, 1998) the level detected in the present study of nucleotide divergence between the ‘lagoon’ and ‘marine’ type populations is higher than those presented in the literature for the intraspecific level and are within the range of 1.4 to 16% for congeneric freshwater fish reported by Billington and Hebert (1991).
There is not always a good correlation between the genetic differentiation revealed by DNA analysis and the morphology displayed by different species (Patterson et al, 1993). Large genetic divergence, at least for some portions of DNA, has already been reported for other groups of fish that have fairly similar morphology (Meyer et al, 1990; Sturmbauer and Meyer, 1992; Patarnello et al, 1994; Papasotiropoulos et al, 2002). This situation could be explained by differences in the selective constraints operating on these two general characters, which seems to lead to the lack of parallel evolution of these characters (Caldara et al, 1996; Rossi et al, 1998).
Although all individuals of A. boyeri populations studied were found to correspond morphologically to the original description, two genetically distinct groups are present, one existing in the ‘marine’ habitat with a second in the ‘lagoon’ habitat. The high levels of genetic differentiation revealed between those two types of populations are also confirmed by the large number of diagnostic patterns existing between them. All these data support the working hypothesis that there are two different sibling species or at least subspecies or semispecies colonizing ‘lagoon’ and ‘marine’ habitats respectively.
Several studies have recently shown that different ecotypes of fishes have experienced rapid genetic divergence and are, in many cases, on the brink of speciation (eg, Taylor et al, 1997; Market et al, 1999). Significant genetic divergence has also been observed between estuarine and nearby marine populations in the silverside fish Odontesthes argentinensis and the authors assume that the colonization of estuarine habitats seems to have promoted rapid adaptive divergence and reproductive isolation in estuarine populations, which were considered as incipient ecological species (Beheregaray and Levy, 2000; Beheregaray and Sunnucks, 2001). Thus the existence of different sibling species in the sea seems not to be a rare phenomenon (for review see Knowlton, 1993). However in our case, further studies with more samples from marine and lagoon habitats are needed to verify our hypothesis.
References
Apostolidis AP, Karakousis Y, Triantaphyllidis C (1996). Genetic differentiation and phylogenetic relationships among Greek Salmo trutta L. (brown trout) populations as revealed by RFLP analysis of PCR amplified mitochondrial DNA segments. Heredity, 77: 608–618.
Avise JC (1991). Ten unorthodox perspectives on evolution prompted by comparative population genetic findings on mitochondrial DNA. Annu Rev Genet, 25: 45–69.
Avise JC (1994). Molecular Markers, Natural History and Evolution. Chapman and Hall: New York.
Bamber RN, Henderson PA (1985). Morphological variation of British atherinids and the status of Atherina presbyter Cuvier (Pisces: Atherinidae). Biol J Linn Soc, 25: 61–76.
Beheregaray LB, Levy JA (2000). Population genetics of the silverside Odontesthes argentinensis (Teleostei, Atherinopsidae); evidence of speciation in an estuary in southern Brazil. Copeia, 2000: 441–447.
Beheregaray LB, Sunnucks P (2001). Fine scale genetic structure, estuarine colonization and incipient speciation in the marine silverside Odontesthes argentinensis. Mol Ecol, 10: 2849–2866.
Bernatchez L, Colombani F, Dodson JJ (1991). Phylogenetic relationships among the subfamily Coregoninae as revealed by mitochondrial DNA restriction analysis. J Fish Biol, 39 (Suppl, A): 283–290.
Berrebi P, Britton-Davidian J (1980). Enzymatic survey of four populations of Atherina boyeri based on electrophoresis and the occurrence of a microsporidosis. J Fish Biol, 16: 149–157.
Billington N, Hebert PD (1991). Mitochondrial DNA diversity in fishes and its implications for introductions. Can J Fish Aquat Sci, 48: 80–94.
Birky CW, Fuerst P, Maruyama T (1989). Organelle gene diversity under migration, mutation, and drift: equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics, 121: 613–627.
Birky CW, Maruyama T, Fuerst P (1983). An approach to population and evolution genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics, 103: 513–527.
Caldara F, Bargelloni L, Ostellari L, Penzo E, Colombo L, Patarnello T (1996). Molecular phylogeny of Grey Mullets based on mitochondrial DNA sequence analysis: Evidence of differential rate of evolution at the intrafamily level. Mol Phylogenet Evol, 6: 416–424.
Cognetti G (1994). Colonization of brackish waters. Mar Pollut Bull, 28: 583–586.
Cognetti G, Maltagliati F (2000). Biodiversity and adaptive mechanisms in brackish water fauna. Mar Pollut Bull, 40: 7–14.
Creech S (1991). An electrophoretic investigation of populations of Atherina boyeri Risso 1810 and A. presbyter Cuvier, 1829 (Teleostei: Atherinidae): genetic evidence in support of the two species. J Fish Biol, 39: 807–816.
Dewoody JA, Avise JC (2000). Microsatellite variation in marine, freshwater and anadromus fishes compared with other animals. J Fish Biol, 56: 461–473.
Farris JS (1977). Phylogenetic analysis under Dollo’s law. Syst Zool, 26: 77–88.
Felsenstein J (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39: 783–791.
Felsenstein J (1993). PHYLIP (Phylogeny Inference Package), Version 3.5C. Distributed by the author. Department of Genetics, University of Washington, Seattle, WA.
Focant B, Rosecchi E, Crivelli AJ (1999). Attempt at biochemical characterization of sand smelt Atherina boyeri Risso 1810 (Pisces, Atherinidae) populations from the Camargue (Rhone delta, France). Comp Biochem Physiol B, 122: 261–267.
Focant B, Trabelsi M, Kartas F, Quignard J-P (1992). Charactérization biochemique de trois populations d’ Atherina boyeri des milineux lagunaires méditerranéens. Rapp Comm Int Mer Medit, 33: 94.
Gon O, Ben-Tuvia A (1983). The biology of Boyeri’s sand smelt Atherina boyeri Risso in the Bardawil Lagoon on the Mediterranean coast of Sinai. J Fish Biol, 22: 537–547.
Henderson PA, Bamber RN (1987). On the reproductive biology of the sand smelt Atherina boyeri Risso, and its evolutionary potential. Biol J Linn Soc, 32: 395–415.
Imsiridou A, Apostolidis AP, Durand JD, Briolay J, Bouvet Y, Triantaphyllidis C (1998). Genetic differentiation and phylogenetic relationships among Greek chub Leuciscus cephalus L. (Pisces, Cyprinidae) populations as revealed by RFLP analysis of mitochondrial DNA. Biochem Syst Ecol, 26: 415–429.
Jackson DP, Hayden JD, Quirke P (1991). Extraction of nucleic acid from fresh and archival material. In McPherson MJ, Quirke P, Taylor GR (eds). PCR, A Practical Approach, IRL Press: Oxford. pp 29–50.
Jones GC, Avise JC (1998). A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochrome b gene. Mol Biol Evol, 15: 1481–1490.
Kartas F, Trabelsi M (1990). Sur le polymorphisme de l'Athérine, Atherina Boyeri Risso 1810 (Atherinidae) des eaux littorales tunisiennes. Cybium, 14: 295–311.
Kiener A, Spillmann CJ (1969). Contribution à l'étude systématique et écologique des Athérines de côtes francaises. Mém Mus Nat Hist Natur Serie A Zoologie, 60: 33–74.
Knowlton N (1993). Sibling species in the sea. Ann Rev Ecol Syst, 24: 189–216.
Lee WJ, Conroy J, Hunting W, Howell N, Kocher TD (1995). Structure and evolution of teleost mitochondrial control regions. J Mol Evol, 41: 54–66.
Lynch M, Crease TJ (1990). The analysis of population survey data on DNA sequence variation. Mol Biol Evol, 7: 377–394.
Market JA, Arnegard ME, Danley PT, Kocher TD (1999). Biogeography and population genetics of the Lake Malawi cichlid Melanochromis auratus: habitat transience, philopatry and speciation. Mol Ecol, 8: 1013–1026.
McElroy D, Moran P, Bermingham E, Kornfield J (1991). The Restriction Enzyme Analysis Package, version 4.0. University of Maine, Orono, ME.
Meyer A, Kocher TD, Basasibwaki P, Wilson AC (1990). Monophyletic origin of Lake Victoria cichlid fishes suggested by mitochondrial DNA sequences. Nature, 347: 550–553.
Page RD (1996). TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci, 12: 357–358.
Palumbi S, Martin A, Romano S, McMillan WO, Stice L, Grabowski G (1991). The simple fools guide to PCR, version II. University of Hawaii, Honolulu.
Papasotiropoulos V, Klossa-Kilia E, Kilias G, Alahiotis S (2002). Genetic divergence and phylogenetic relationships in grey Mullets (Teleostei: Mugilidae), based on PCR-RFLP analysis of mtDNA segments. Biochem Genet, 40: 71–86.
Patarnello T, Bargelloni L, Caldara F, Colombo L (1994). Cytochrome b and 16S rRNA sequence variation in the Salmo trutta (Salmonidae, Teleostei) species complex. Mol Phylogenet Evol, 3: 69–74.
Patterson C, Williams DM, Humphries CJ (1993). Congruence between molecular and morphological phylogenies. Annu Rev Ecol Syst, 24: 153–188.
Roff DA, Bentzen P (1989). The statistical analysis of mitochondrial DNA polymorphisms: χ2 and the problem of small samples. Mol Biol Evol, 6: 539–545.
Rossi AR, Capula M, Crosetti D, Campton DE, Sola L (1998). Genetic divergence and phylogenetic inferences in five species of Mugilidae (Pisces: Perciformes). Mar Biol, 131: 213–218.
Sneath PH, Sokal RR (1973). Numerical taxonomy – The principles and practice of numerical classification. Editors Freeman WH and Co: San Francisco.
Sturmbauer C, Meyer A (1992). Genetic divergence, speciation and morphological stasis in a lineage of African cichlid fishes. Nature, 358: 578–581.
Taylor EB, Harvey S, Pollard S, Volpe J (1997). Postglacial genetic differentiation of reproductive ecotypes of kokanee Oncorynchus nerka in Okanagan Lake, British Columbia. Mol Ecol, 6: 503–517.
Trabelsi M, Quignard J-P, Kartas F (1994). Atherina boyeri: première mention en Méditerranée de deux populations marines sympatriques. Cybium, 18: 457–459.
Triantafyllidis A, Abatzopoulos TJ, Economidis PS (1999). Genetic differentiation and phylogenetic relationships among Greek Silurus glanis and Silurus aristotelis (Pisces, Siluridae) populations, assessed by PCR-RFLP analysis of mitochondrial DNA segments. Heredity, 82: 503–509.
Wilson AC, Cann RL, Carr SM, George M, Gyllensten UB, Helm-Bychowski KM et al. (1985). Mitochondrial DNA and two perspectives on evolutionary genetics. Biol J Linn Soc, 26: 376–397.
Acknowledgements
We are grateful to Dr P Kaspiris for helping us in the morphological classification of the specimens as well as the two anonymous referees for their helpful criticism. A part of this work was supported by a grant from the research Committee of Patras University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Klossa-Kilia, E., Prassa, M., Papasotiropoulos, V. et al. Mitochondrial DNA diversity in Atherina boyeri populations as determined by RFLP analysis of three mtDNA segments. Heredity 89, 363–370 (2002). https://doi.org/10.1038/sj.hdy.6800144
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800144
Keywords
This article is cited by
-
Biogeographical history of the genus Atherina (Pisces: Atherinidae) in the central Eastern Atlantic
Marine Biodiversity (2019)
-
Genetic differentiation among Greek lake populations of Carassius gibelio and Cyprinus carpio carpio
Genetica (2009)
-
Genetic Divergence Among Marine and Lagoon Atherina boyeri Populations in Greece Using mtDNA Analysis
Biochemical Genetics (2008)
