
Abstract
Family-based studies to map susceptibility genes through linkage disequilibrium have been successful in early-onset diseases where parental-proband trios are readily collected, but are believed to be unworkable for late-onset diseases such as coronary artery disease (CAD). PROCARDIS is a European multicentre study that was designed to identify susceptibility genes for CAD. We have tested the transmission of a putatively functional allele, lymphotoxin-α N26 (804A), in more than 400 PROCARDIS trio families. The present study demonstrates association of this allele with CAD in white Europeans, a different ethnic group with a heavier CAD burden than the Japanese in which the association was initially identified, which suggests a broad relevance to CAD susceptibility. The practicalities of implementing a trio-family design for late-onset diseases are discussed.
Similar content being viewed by others

Introduction
The PROCARDIS study is a European collaborative project that is using genetic techniques to map susceptibility genes for coronary artery disease (CAD). In the first phase of this research, we have set up a clinical network to recruit families to implement two study designs based on linkage and transmission disequilibrium test (TDT) analyses. We have investigated a haplotype, reported to be associated with myocardial infarction (MI) in Japanese, to test the feasibility of the TDT approach in the late-onset disease CAD.
In a recent large-scale ‘genome-wide’ association study, 66 000 largely gene-based SNPs were tested for association with MI in a two-stage case–control study that included a total of 1133 Japanese patients.1 Polymorphisms in the vicinity of the lymphotoxin-α gene (LTA) on chromosome 6p21 showed the strongest evidence of association; haplotype analysis suggested that the association extended in a block over approximately 50 kb and included SNPs within the NFKBIL1 gene (encoding nuclear factor of κ light polypeptide gene enhancer in B cells, inhibitor-like 1) and the BAT1 gene (encoding HLA-B-associated transcript 1). The candidature of several SNPs was assessed in in vitro functional studies and highlighted the potential causative role of several of these SNPs as heritable risk factors for myocardial infarction: 252G in intron 1 of LTA was associated with higher transcription and stronger binding of a nuclear factor than 252A;1, 2, 3, 4 recombinant human N26-LTA induced more VCAM1 and SELE mRNA production from cultured human coronary artery smooth muscle cells than T26-LTA;1 the −63A allele of the NFKBIL1 promoter, on the same haplotype as LTA 252G and N26, showed slightly lower transcriptional activity.1
We have tested the postulated association of LTA with CAD in PROCARDIS trio families, who differ from the Japanese in their ‘genetic ancestry’. Moreover, the rates of CAD are approximately five-fold lower in Japan than in white European populations such as PROCARDIS, which may in part reflect the different genetic loads present in each population.
Materials and methods
Ascertainment criteria for PROCARDIS probands were MI or symptomatic acute coronary syndrome (SACS; on the assumption that the latter represents a similar pathological process) according to modified WHO diagnostic criteria,5, 6 before the age of 65 years. Briefly, diagnosis of MI required documentation of two or more of: (a) typical ischaemic chest pain, pulmonary oedema, syncope or shock; (b) development of pathological Q-waves and/or appearance or disappearance of localized ST elevation followed by T-wave inversion in two or more standard electrocardiograph leads; (c) increase in concentration of serum enzymes consistent with MI (eg creatine kinase more than twice the upper limit of normal). Diagnosis of SACS required documentation on hospitalization for one of the following indications: (a) unstable angina diagnosed by typical ischemic chest pain at rest associated with reversible ST depression in two or more standard electrocardiograph leads; (b) thrombolysis for suspected MI (as indicated by localized ST elevation in two or more standard electrocardiograph leads) even without later development of T-wave inversion, Q-waves or a significant enzyme rise; (c) emergency revascularization (ie during same admission) following presentation with typical ischaemic chest pain at rest.
For the TDT study design, probands completed a questionnaire to establish the availability of their parents and siblings. Both parents whenever possible, or one parent and at least one sibling (affected or unaffected), were then invited to participate, along with any further affected siblings conforming to the PROCARDIS proband definition. Full ethical approval was obtained and all participants gave written informed consent. Families were recruited in four countries: in Sweden and the UK, recruitment was based on existing databases of patients hospitalized for CAD; in Sweden for a few families and in Germany for all, recruitment was through lists of MI patients currently or recently admitted to coronary care units (CCUs); in Italy, recruitment was through both MI patient databases and CCUs.
We extracted DNA from blood using standard techniques. We determined genotype for the lymphotoxin-α T/N26 (804A/C) SNP using the high-throughput PCR-based Sequenom® technology that uses primer extension and detects absolute allele-specific mass differences using MALDI-TOF mass spectrometry. The PCR primers were 5′ACGTTGGATGTCAGCACCCCAAGATGCATC3′ and 5′ACGTTGGATGGAGGTCAGGTGGATGTTTAC3’ and the extension primer was 5′GATGCATCTTGCCCACAGCA3′; an extension/termination mix containing dATP, ddCTP, ddGTP and ddTTP was used.
The programme PedCheck7 was used to check for misinheritance using the full database of 44 SNPs and three microsatellites genotyped on these families; individuals showing more than two instances of misinheritance were excluded, which in some cases led to the exclusion of entire families. Excess transmission of the LTA N26 allele to the affected offspring was tested using two statistical methods that amalgamate information from complete trios (where genotypes are available for both parents and the proband) and incomplete trios (where genotypes are available from the proband, one parent and one or more additional siblings). First, we used the computer programme Transmit (version 2.58) that implements a score-test function that omits the terms that are most affected by population stratification; the robust variance estimation option was selected to allow for the nonindependence in families with more than one affected sib. We tested for segregation distortion by analysing transmission to offspring not known to be affected, defined as having no self-reported CAD history or symptoms. Secondly, we used the Pedigree Analysis Package (PAP version 5.0, http://hasstedt.genetics.utah.edu), which implements a full-likelihood solution which may not be robust to departures from a panmictic, unstructured population model;9 maximum likelihood and standard error estimates of genetic parameters were obtained numerically using the GEMINI subroutine.
Results
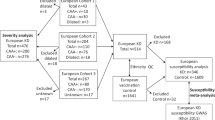
For this PROCARDIS TDT study, we recruited 460 trio families, comprising 131 with proband and both parents and 329 with proband plus one parent and at least one sibling. PedCheck analysis of all the available genotype data revealed several instances of misinheritance that led to the complete exclusion of 13 families, making a total of 447 families (117 with both parents). The families originated in the following countries (families with both parents shown in parenthesis): 55 (16) from Germany, 255 (56) from Italy, 85 (35) from Sweden and 52 (10) from the UK. Although we took MI or SACS as criteria for affected status, the vast majority of the affected offspring had suffered MI (Germany 90%, Italy 88%, 97% Sweden, 98% UK).
Five LTA SNPs in the linkage disequilibrium (LD) block implicated in CAD in the Japanese1 (Table 1) were confirmed to be polymorphic in our four white European populations. Clark's algorithm10 was used to derive haplotypes for the five SNPs shown in Table 1 in 23 PROCARDIS probands. This resulted in the unambiguous assignment of two haplotypes only, inferring complete LD (data not shown), and mirroring the findings in the Japanese study. We selected one of these SNPs, the lymphotoxin-α T/N26 (804A/C), for genotyping in the PROCARDIS families as it showed the strongest association in the study by Ozaki et al,1 who suggested that it may be functional.
We analysed the LTA T/N26 (804A/C) SNP in 447 trio families with 474 affected offspring that gave 303 informative transmissions. The frequency of the N26 allele was 0.30 (95% confidence interval 0.27–0.33), which is slightly lower than the frequency of 0.37 (0.34–0.40) in the Japanese controls and 0.41 (0.38–0.44) in the Japanese myocardial infarction cases.1 Using the Transmit programme, there was evidence for excess transmission of the LTA N26 allele to affected offspring, with χ2 (1 d.f.)=8.44 and P=0.0018 (a one-tailed test is appropriate as we are testing the transmission of a specific allele). The equivalent number of fully informative (ie heterozygous parents) transmissions was estimated as 303 (calculated by multiplying the variance of the ‘observed–expected’ score statistic by four). A weighted analysis, using reciprocal-variance weights, suggested that there was no difference in the strength of the gene-association across the four countries (heterogeneity test statistic=1.4, 3 d.f., P=0.7), thus confirming the strong evidence of gene-association, with χ2 (1 d.f.)=8.47 and P=0.0018. We also tested the transmission of the N26 (804A) allele to CAD offspring who declared themselves to be unaffected (status not verified, see Materials and methods; 288 equivalent informative transmissions) and found no excess transmission with χ2 (1 d.f.)=2.07, P=0.08 and τ=0.46 (0.40–0.52), suggesting that this allele is not subject to segregation distortion.
The proportion of N26 (804A) alleles transmitted to affected individuals was estimated as 0.58 (0.53–0.64) using PAP. There is a simple relationship (under a multiplicative model of allele risk) between this transmission proportion (τ) and the genotype risk ratio (γ)11 such that γ=τ/(1−τ) which, under assumptions of Hardy–Weinberg equilibrium, is equivalent to the odds ratio (OR) calculated in a case–control study. The genotype risk ratio for the N26 allele from the PROCARDIS trio families was thus calculated to be 1.40 (1.12–1.77). Ozaki et al found a recessive association of the N26 allele with MI, so to make a direct comparison, the PROCARDIS genotype risk ratio for N26 homozygotes, γ2, was calculated to be 1.96 (1.25–3.13). This is comparable with the OR of 1.78 (1.39–2.27) found in the Japanese population.1
Discussion
Several studies have suggested that the LTA 252G allele is associated with increased transcription,1, 2, 3, 4 whereas the NFKBIL1 −63A allele caused a moderate lowering of transcription. Ozaki et al1 also showed that recombinant LTA with asparagine at amino acid 26 stimulated almost double the production of VCAM1 and E-Selectin mRNAs in cultured human coronary artery smooth muscle cells compared with recombinant LTA with threonine at position 26, and so appears strongly proinflammatory. Using a novel technique to quantitate allele-specific transcriptional activity at any given genomic position, Knight et al3 were able to show that the LTA 252G-containing haplotype (denoted haplotype A) was more transcriptionally active than another haplotype denoted B, but were unable to distinguish it from a third haplotype C in terms of transcriptional activity. This A or ‘active’ haplotype corresponds to the A haplotype identified by Ozaki et al, the B haplotype to haplotypes B and C identified by Ozaki et al, and C to D and E, although only haplotype A showed association with MI in the Japanese population-based study. Clearly functional data should be instructive for dissecting the mechanism of action of these SNPs, but obtaining the correct experimental result depends on using the appropriate cell type and stimulus in such in vitro experiments. Therefore, it remains essential to identify disease-causing haplotypes by genetic methods.
Our results, together with the Japanese case:control study, suggest that it is the 252G/N26-containing haplotype that is important in vivo in the MI/CAD association. The extent of the LD in this region is known to be high, but Knight et al have shown convincingly that the association with higher transcriptional activity of the LTA 252G allele is not due to LD with the neighbouring TNFα -308G/A SNP, which appears nonfunctional under the conditions tested.3 It remains to be seen whether the LTA 252G/N26 functional haplotype extends further, and whether other individual SNPs in the haplotype are also functional, but the present study strongly reinforces the contention that this haplotype, defined by at least three functional SNPs, may be causally related to MI and/or CAD. A very recent case:control study of MI, also in Japanese, confirmed association of the LTA 10A/252G/N26-containing haplotype with increased risk of MI, although in this study the effect was dominant, with an OR of 1.7 (1.2–2.3) for presence of the LTA10A allele (10A/252G/N26-haplotype), which is comparable with our genotype risk ratio for the presence of the same haplotype (tagged using the T/N26 SNP) of 1.40 (1.12–1.77).12
Both the present study and the Japanese case:control studies were retrospective, therefore, it remains a possibility that the LTA N26 haplotype is a risk factor only in survivors of MI and/or CAD and it would be interesting to investigate this relationship in a prospective setting, perhaps in a nested case:control study.
There has been much discussion recently as to the relative merits of transmission-based tests in trio families versus testing for association in case:control series. Although some investigators have concluded that the problem of population stratification in case:control studies may have been overstated,13 recent data suggest that it may yet remain a significant problem14, 15 and so freedom from concerns regarding stratification remains attractive. We note that the Japanese control genotype frequencies for LTA 804C/A were not in Hardy–Weinberg equilibrium, which may have inflated the significance of their findings as some level of admixture may have contributed to this observation.16 It is also pertinent to note that collection of true negative controls is difficult in CAD as presymptomatic disease will be common in apparently healthy control individuals. In contrast, use of trio families does not require any control population, thus avoiding any such distortion of results. A significant proportion of genotyping errors can be eliminated in trios if they result in Mendelian segregation inconsistencies;17 this is important as such errors have a negative impact on the power of gene-association studies.18 There are concerns that for dominant diseases, excess mortality in the ‘carrier’ parent will lead to biases.19 The corollary, however, is that families carrying recessive alleles will be preferentially sampled in trio cohorts and consequently as both parents are informative, the power will be optimal in the TDT design. Additionally, family-based studies uniquely permit parent-of-origin tests.20 However, overriding all these statistical genetics issues, it is widely believed that it would not be possible to collect sufficient numbers of trio families in a complex, late-onset disease like CAD.21, 22 By contrast, the present results show that it is possible not only to collect sufficient informative families but also to use them to detect the modest genotype risk ratios expected for such a complex disease. Our success relied on the close coupling of the trio collection with an affected sibling pair collection framework; setting up de novo a project to ascertain and collect trios alone for late-onset diseases would be difficult to justify given prevailing trends.
In conclusion, we have shown strong linkage/association of the LTA haplotype identified by the 804A (N26) allele with CAD, confirming the findings of one of the first genome-wide association studies in a complex trait. As this finding was replicated in a different ethnic group, our data support the likely functional role of the LTA N26 allele in CAD. Finally, we have demonstrated the utility of a complementary experimental design, the trio family approach.
References
Ozaki K, Ohnishi Y, Iida A et al: Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 2002; 32: 650–654.
Messer G, Spengler U, Jung MC et al: Polymorphic structure of the tumor necrosis factor (TNF) locus: an NcoI polymorphism in the first intron of the human TNF-beta gene correlates with a variant amino acid in position 26 and a reduced level of TNF-beta production. J Exp Med 1991; 173: 209–219.
Knight JC, Keating BJ, Rockett KA, Kwiatkowski DP : In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nat Genet 2003; 33: 469–475.
Whichelow CE, Hitman GA, Raafat I, Bottazzo GF, Sachs JA : The effect of TNF*B gene polymorphism on TNF-alpha and -beta secretion levels in patients with insulin-dependent diabetes mellitus and healthy controls. Eur J Immunogenet 1996; 23: 425–435.
ISFC/WHO: Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology/World Health Organization task force on standardization of clinical nomenclature. Circulation 1979; 59: 607–609.
Gillum RF, Fortmann SP, Prineas RJ, Kottke TE : International diagnostic criteria for acute myocardial infarction and acute stroke. Am Heart J 1984; 108: 150–158.
O'Connell JR, Weeks DE : PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63: 259–266.
Clayton D : A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 1999; 65: 1170–1177.
Monks SA, Kaplan NL, Weir BS : A comparative study of sibship tests of linkage and/or association. Am J Hum Genet 1998; 63: 1507–1516.
Clark AG : Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 1990; 7: 111–122.
Risch N, Merikangas K : The future of genetic studies of complex human diseases. Science 1996; 273: 1516–1517.
Iwanaga Y, Ono K, Takagi S et al: Association analysis between polymorphisms of the lymphotoxin-alpha gene and myocardial infarction in a Japanese population. Atherosclerosis 2004; 172: 197–198.
Cardon LR, Palmer LJ : Population stratification and spurious allelic association. Lancet 2003; 361: 598–604.
Freedman ML, Reich D, Penney KL et al: Assessing the impact of population stratification on genetic association studies. Nat Genet 2004; 36: 388–393.
Marchini J, Cardon LR, Phillips MS, Donnelly P : The effects of human population structure on large genetic association studies. Nat Genet 2004.
Deng HW, Chen WM, Recker RR : Population admixture: detection by Hardy–Weinberg test and its quantitative effects on linkage-disequilibrium methods for localizing genes underlying complex traits. Genetics 2001; 157: 885–897.
Geller F, Ziegler A : Detection rates for genotyping errors in SNPs using the trio design. Hum Hered 2002; 54: 111–117.
Gordon D, Finch SJ, Nothnagel M, Ott J : Power and sample size calculations for case–control genetic association tests when errors are present: application to single nucleotide polymorphisms. Hum Hered 2002; 54: 22–33.
Curtis D : Use of siblings as controls in case–control association studies. Ann Hum Genet 1997; 61 (Part 4): 319–333.
Weinberg CR : Methods for detection of parent-of-origin effects in genetic studies of case–parents triads. Am J Hum Genet 1999; 65: 229–235.
Morton NE, Collins A : Tests and estimates of allelic association in complex inheritance. Proc Natl Acad Sci USA 1998; 95: 11389–11393.
Cardon LR, Bell JI : Association study designs for complex diseases. Nat Rev Genet 2001; 2: 91–99.
Acknowledgements
We gratefully acknowledge grants from AstraZeneca and the European Commission.
Author information
Authors and Affiliations
Consortia
Rights and permissions
About this article
Cite this article
The PROCARDIS Consortium. A trio family study showing association of the lymphotoxin-α N26 (804A) allele with coronary artery disease. Eur J Hum Genet 12, 770–774 (2004). https://doi.org/10.1038/sj.ejhg.5201244
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201244
Keywords
This article is cited by
-
Multiple selective sweeps of ancient polymorphisms in and around LTα located in the MHC class III region on chromosome 6
BMC Evolutionary Biology (2019)
-
Molecular genetics of coronary artery disease
Journal of Human Genetics (2016)
-
Variants at the Endocannabinoid Receptor CB1 Gene (CNR1) and Insulin Sensitivity, Type 2 Diabetes, and Coronary Heart Disease
Obesity (2011)
-
A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease
Nature Genetics (2011)
-
Leprosy as a genetic disease
Mammalian Genome (2011)
