
Abstract
The hypoxia-inducible factors (HIFs) are critical for cellular adaptation to limiting oxygen and regulate a wide array of genes when cued by cellular oxygen-sensing mechanisms. HIF is able to direct transcription from either of two transactivation domains, each of which is regulated by distinct mechanisms. The oxygen-dependent asparaginyl hydroxylase factor-inhibiting HIF-1α (FIH-1) is a key regulator of the HIF C-terminal transactivation domain, and provides a direct link between oxygen sensation and HIF-mediated transcription. Additionally, there are phosphorylation and nitrosylation events reported to modulate HIF transcriptional activity, as well as numerous transcriptional coactivators and other interacting proteins that together provide cell and tissue specificity of HIF target gene regulation.
Similar content being viewed by others

Main
The maintenance of oxygen homeostasis is a crucial physiological requirement that involves coordinated regulation of a plethora of genes. The hypoxia-inducible transcription factors (HIFs) are responsible for a major genomic response to hypoxia, where cellular oxygen demand exceeds supply. The HIFs directly regulate the transcription of more than 70 genes involved in cellular processes that act to directly address this deficit by decreasing oxygen dependence and consumption by cells, and by increasing the efficiency of oxygen delivery to cells. These processes include vasculogenesis and angiogenesis, metabolism, vasodilation, cell migration, signalling and cell fate decisions. As such, the HIFs are fundamental to embryonic development and the pathophysiology of many serious human diseases, and are therefore subject to strict regulatory mechanisms that act to limit transcriptional activity to periods of cellular oxygen stress. The HIF proteins are subject to oxygen-dependent regulation, both at the level of protein stability (reviewed in this issue by R Bruick) and transcriptional activity, rendering them almost inactive at normoxia, but potently inducible in hypoxia. The regulation of the transcriptional activity of the HIF proteins, both via oxygen-dependent and oxygen-independent mechanisms, will be discussed in this review.
The HIFs
HIF is assembled from α- and β-subunits to become a transcriptionally active heterodimer.1 Both subunits are members of the bHLH/PAS (basic helix–loop–helix/Per-Arnt-Sim homology) family of transcription factors, and both contain transactivation domains (Figure 1).2, 3 Whereas HIF-1β, also known as the aryl hydrocarbon receptor nuclear translocator (Arnt1), is a constitutively expressed nuclear protein, the α-subunit is strictly regulated in an oxygen-dependent manner. There are three paralogues of the HIF-α subunit, HIF-1α, HIF-2α and HIF-3α, and three paralogues of HIF-1β (Arnt1, Arnt2 and Arnt3), with either HIF-1α or HIF-2α being able to heterodimerize with HIF-1β to form the functional HIF transcription factor complexes responsible for the hypoxic shift in gene expression. HIF-3α has no known role as an active transcription factor, and is instead postulated to behave as a negative regulator of HIF-mediated transcription.4 An alternative splicing product of the HIF-3α gene has been found to dimerise with HIF-1β and inhibit transcription of target genes by sequestration of HIF-1β from HIF-α.5 The alternative splicing event leading to generation of this factor, termed inhibitory PAS domain protein (IPAS), is hypoxically inducible, and IPAS may therefore provide a potential negative feedback mechanism for HIF-α regulation.6, 7

Domain structures of the HIF transcription factors. The three HIF-α paralogues, HIF-1α, HIF-2α and HIF-3α (including the HIF-3α splice variant IPAS), and common binding partner HIF-1α or Arnt, all contain bHLH and PAS domains. The three HIF-α isoforms all contain ODDDs, NADs and CADs. HIF-1α and HIF-2α each contain an oxygen-regulated CAD. Numbers refer to amino-acid similarity between human HIF-1α and HIF-2α in the defined domains2
DNA Binding
HIF-α is constitutively transcribed and translated in cells, but under normoxia it has an extremely short half-life of less than 5 min.8, 9 This rapid normoxic turnover is facilitated by hydroxylation of specific proline residues within a central oxygen-dependent degradation domain (ODDD; Figure 1) by HIF-α-specific prolyl hydroxylases (PHDs, reviewed in this issue by R Bruick). Hydroxylation prompts polyubiquitylation of HIF-α, targeting it for proteasomal degradation. Hypoxia, however, impedes hydroxylation and stabilises the protein. Regulation of HIF-α stability is also mediated by an oxygen-independent pathway. In the cytoplasm, HIF-α is bound by heat-shock protein 90 (Hsp90) and this association leads to enhanced stability of the subunit.10 However, upon displacement of Hsp90 by Hsp90 inhibitors, receptor of activated protein C kinase (RACK1) is able to bind and acts to recruit ubiquitin ligase machinery and potentiate the degradation of the α-subunit.
When stable, HIF-α translocates to the nucleus, dimerises with HIF-1β and binds to hypoxia response elements (HREs) in the regulatory regions of target genes. Dimerisation is an absolute prerequisite for DNA binding and is mediated by the bHLH and PAS domains of each subunit, with the basic regions contiguous with the HLH motifs of both partners contacting the DNA (Figure 2).11 Dissection of the enhancer of the seminal HIF target erythropoietin (EPO)12 revealed two regions essential for reporter activity; the core HRE or HIF-binding site (HBS) containing the consensus sequence (A/G)CGTG) which HIF contacts directly, and the HIF ancillary sequence (HAS), which is an imperfect inverted repeat shown to recruit transcription factor complexes other than HIF.13 Over 70 genes have been confirmed as bona fide HIF targets containing the canonical HBS. Although microarray experiments indicate that more than 200 transcripts are hypoxically regulated, some of these may be indirect targets or regulated in an HIF-independent manner.14

HIF dimerisation and DNA binding. Representation of the bHLH domains of the HIF-α/β heterodimer contacting DNA. The basic residues near the N-terminus of each protein contact the nucleotides of the core HRE directly to form an α-helix contiguous with helix 1
HIF Transactivation Domains
Once HIF is assembled on the HRE, it must recruit transcriptional coactivators to form an intact initiation complex, a process mediated by distinct transactivation domains present within the HIF-α proteins. HIF-1α and HIF-2α contain two transactivation domains, an oxygen-regulated C-terminal transactivation domain (CAD, spanning residues 786–826 of hHIF-1α) and a more centrally located transactivation domain designated N-terminal transactivation domain (NAD, spanning residues 531–575 of hHIF-1α; Figure 1).15, 16 The NAD and CAD of HIF-1α are each highly conserved between species, with more than 90 and 100% amino-acid conservation, respectively, between mice and humans. However, the NAD and CAD of human HIF-1α share little amino-acid similarity with each other, implying divergent yet highly important roles for each domain.17
Both the NAD and CAD employ recruitment of the coactivators CBP/p300, SRC-1, and transcription intermediary factor 2 (TIF-2),18, 19, 20 although direct interactions have only been demonstrated between the CAD and p300 or Creb-binding protein (CBP).21 CBP and p300 are paralogous transcriptional coactivators that are essential for linking HIF and other transcription factors with coactivator complexes and the basal transcriptional machinery, and are thus indispensable for robust transcriptional activation. Additionally, CPB/p300 have histone acetyltransferase activity that is needed for modification of chromatin prior to transcription. Although the obligate partner HIF-1β has its own well-defined CAD (Figure 1), it appears to be dispensable for transcription in the context of the HIF heterodimer.2 However, it remains a possibility that other uncharacterised transactivation domains exist within HIF-1β, and they are required for HIF-mediated transcription.22
A key difference between the NAD and the CAD is their oxygen-dependent regulation. The central NAD is contiguous with the ODDD, and thus it has been difficult to distinguish NAD-specific regulation from the oxygen-dependent protein degradation mediated by the ODDD.16 However, although there have been some reports suggesting some oxygen-dependent regulation of the NAD independent of stability, any such regulation is minor compared with the highly regulated CAD.15
A recent study has characterised the differential roles of NAD- and CAD-driven transcription.17 By manipulating the levels of the hydroxylases within cells to modulate NAD and CAD activity, it was concluded that the CAD contributed to the regulation of most, but not all, HIF target genes, and was the predominant transactivation domain. However, a subset of HIF target genes that dependent exclusively on the NAD and not influenced by changes in CAD activity, supporting the specific roles of the two HIF transactivation domains. Physiologically, these genes are hypothesised to be activated in response to oxygen concentrations below the threshold required for PHD activity, yet still adequate for factor-inhibiting HIF (FIH) to hydroxylate and therefore silence the CAD.
The NAD has recently been shown to contribute to HIF-1 and HIF-2 target gene specificity.23 HIF-1α/HIF-2α chimeric proteins, where innate NAD and/or CAD domains of each paralogue were ‘swapped’, were used to investigate the effect of each on the transcription of endogenous HIF target genes deemed to be either HIF-1α specific, common targets of both HIF-1α and HIF-2α, or HIF-2α specific. Interestingly, swapping the CAD between HIF-1α and HIF-2α had no effect on the target transcripts measured, whereas substitution of both the NAD and the CAD effectively caused HIF-1α to behave as HIF-2α, and vice versa. These data clearly implicate the NAD and not the CAD in regulating HIF target gene specificity.
In isolation, the 40-amino-acid CAD is constitutively active in reporter gene assays independent of oxygen levels. However, when the adjacent inhibitory domain (ID) is included (located between the two transactivation domains; Figure 1), the CAD is efficiently repressed in normoxia and derepressed in hypoxia.15 A more detailed characterisation of the ID demonstrated that as little as 10 amino acids adjacent to the CAD are required for normoxic repression, and alanine scanning mutagenesis showed the importance some or all of the RLL triad located five residues N-terminal to the CAD for this repression.24 Regulation of CAD activity has been of considerable interest, as this involves the second oxygen-dependent posttranslational modification regulating HIF.
Oxygen-Dependent Regulation of HIF-CAD by FIH-1
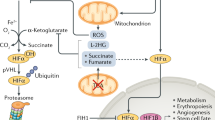
Whereas stability of HIF-α is regulated by the hydroxylation of two conserved prolyl residues within the ODDD by three HIF prolyl hydroxylases (PHD 1–3), the activity of the CAD is regulated by hydroxylation of a single conserved asparaginyl residue by the asparaginyl hydroxylase, FIH-1.21, 25, 26, 27 FIH-1, like the PHDs, is a member of the Fe(II) and 2-oxoglutarate (2-OG)-dependent dioxygenase family of hydroxylases. FIH-1 catalyses the splitting of dioxygen, using one oxygen atom for hydroxylation of the target asparagine, and the other for oxidising the cofactor 2-OG to succinate, thus releasing CO2 (Figure 3).

Normoxic silencing of the CAD by FIH-1-mediated hydroxylation. At normoxia, FIH uses the co-substrates O2 and 2-OG to hydroxylate the target asparagine residue within the HIF-α-CAD, releasing CO2 and succinate. This hydroxylation event precludes association with the essential coactivators CBP/p300, repressing the transcriptional activity of the CAD. During hypoxia, when oxygen is limiting, FIH is unable to efficiently catalyse the hydroxylation of the CAD, enabling binding of CBP/p300 to the non-hydroxylated CAD and transactivation of target genes
The oxygen-dependent hydroxylation of this single asparagine was first identified in mHIF-2α at position 851, corresponding to Asn803 in hHIF-1α.21, 26, 27 Hydroxylation at this single asparagine residue was found to be sufficient to prevent the interaction of the CAD with the essential transcriptional coactivator CBP/p300, thus silencing HIFs transcriptional ability.25, 28 Replacement of the target asparagine with alanine renders the CAD constitutively active and capable of binding the CBP/p300 coactivator complex irrespective of O2 levels, showing that asparagine hydroxylation is necessary for CAD repression at normoxia.
Hypoxia inhibits CAD hydroxylation by inhibiting FIH-1 activity, facilitating CBP/p300 recruitment and providing a direct mechanism for the hypoxic activation of the CAD. Thus, modification of the target asparagine acts as a molecular switch, enabling repression of the CAD at normoxia, and derepression in hypoxia when HIF target genes are required by the cell.21
Oxygen-Dependent Regulation of FIH-1
FIH-1 has an absolute requirement for oxygen to maintain enzymatic activity, and thus has the potential to be a cellular oxygen sensor. The estimated Km values of FIH-1 and the PHDs for molecular oxygen were first reported to be approximately 90 and 250 μM, respectively.29, 30 Although these values were determined in vitro using relatively short peptides as substrates, they do provide a guide as to the oxygen sensitivity of these enzymes within cells. Given that physiological oxygen concentrations are around 4–40 μM and below the estimated Km, and the enzymes are present within cells in limiting amounts, this implies that the hydroxylases are poised to respond to physiologically relevant changes in intracellular oxygen concentrations, with any decrease in oxygen tension likely to impart a direct reduction in the catalytic rate of the enzymes (reviewed in Schofield and Ratcliffe31and Fandrey et al.32). Although these data are in support of FIH-1 and the PHDs acting as direct cellular oxygen sensors, it is also possible that these hydroxylases are indirect oxygen sensors, with oxygen sensing mediated by mitochondria, for example, via generation of reactive oxygen species in hypoxia,33 although this remains controversial.
Interestingly, since FIH-1 appears to have a higher affinity for O2 than the PHDs based on in vitro experiments,29 it follows that as the severity of hypoxia increases, the PHDs would be inactivated first, while FIH-1 would require more severe hypoxia to lose activity (Figure 4). Indeed, overexpressed FIH-1 can still exert a catalytic effect at 0.2% oxygen concentration, whereas the PHDs were found to be inactive under the same conditions.34 However, more recent studies using larger peptides suggest that the Km of FIH-1 for oxygen in vitro is closer to 250 μM like the PHDs, depending on the length of peptide used.35 More importantly, cell-based assays examining the activity of endogenous FIH-1 and the PHDs indicate that FIH-1 was less sensitive to decreasing oxygen levels than the PHDs in some cell types, whereas in other cells types it was more sensitive, thus demonstrating that the oxygen-dependent sensitivity of FIH-1 can vary between different cell types independently of the PHDs.36 These cell-based results do not invalidate the in vitro results, but rather reflect the intrinsic differences between proteins in a complex cellular environment compared with isolated proteins and peptides.

Hypothesised sequential activation of the NAD, and then the CAD, in gradients of hypoxia. Predicted from in vitro derived Km values of PHD and FIH for oxygen, this model suggests that as oxygen concentrations decrease, the PHDs lose catalytic activity first due to their weaker affinity for O2, while FIH still hydroxylates and represses the CAD. This leads to stabilisation of only HIF-α protein and NAD activity. At lower oxygen tensions, catalytic activity of FIH-1 is reduced, derepressing the CAD for full transcriptional activation
The Structure of HIF–CAD
Determination of the structure of HIF-α–CAD when in complex with either CBP/p300 or FIH-1, has contributed to understanding the inherently malleable nature of the CAD, which adopts different structures depending on the nature of its binding partner, and helped understand the mechanism by which hydroxylation of Asn803 abrogates CBP/p300 binding.
First, structural analysis by nuclear magnetic resonance showed that the unbound hHIF-1α–CAD (residues 776–826) is unstructured.37, 38 Upon binding FIH-1, the CAD becomes structured and engages the FIH-1 homodimer at two sites of interaction; site 1 in hHIF-1α–CAD encompasses residues 795–806 and contains the hydroxylated asparagine, while site 2 includes residues 812–823 and shows only weak binding independent of site 1 (Figure 5).39 Site 1 binds via 10 hydrogen bonds in an extended loop conformation, and site 2 residues form an α-helix.39 In addition, FIH-1 itself undergoes a conformational change upon HIF–CAD binding.

Induced-fit model of HIF-α-CAD (residues 775–826) binding. The unbound CAD is unstructured. It is able to bind to either FIH or CBP/p300, via two distinct sites of interaction (site 1, residues 795–806; and site 2, residues 813–822), and to adopt different conformations at these sites depending on the binding partner. While the α-helical conformation at site 2 is similar when bound to FIH or CBP/p300, the α-helix at site 1 is only observed when it is bound to CBP/p300
Within the FIH-1–CAD complex, Asn803 is deeply embedded within the catalytic pocket, guided by a hydrogen bond between the backbone carbonyl group of Val802 and the backbone amino group of Ala804. This interaction ratifies the finding that after individual mutagenesis of eight conserved residues of the HIF-1α–CAD close to Asn803, only the adjacent Val802 was found to be essential for efficient hydroxylation.40 Mutation of this residue to alanine reduced in vitro catalysis of hydroxylation fourfold, without interrupting substrate binding, a result corroborated in mammalian cell-based reporter assays.40 Recognition and hydroxylation of the CAD by FIH-1 have also been reported to require a minimal substrate sequence of approximately 35 residues, including residues 788–822, which essentially spans sites 1 and 2.29 Hydroxylation is markedly reduced by deletion of only a few residues from either the N- or C-terminus, demonstrating the importance of both site 1 and site 2 in FIH-1 binding and hydroxylation.
The CAD interacts with CBP/p300 via the cysteine/histidine-rich domain 1 (CH1 domain) of the latter, which comprises four α-helices that form a hydrophobic core. In contrast to the extended loop formed by hHIF-1α–CAD site 1 residues when interacting with FIH-1, upon binding to the CH1 domain, both site 1 and site 2 residues adopt an α-helical conformation (helices spanning residues 797–803 and 816–822) (Figure 5), connected by an extended loop that straddles helix 3 of the CH1 domain.37, 38 Within the hHIF-1α–CAD/p300 complex, the side chain of Asn803 is the most deeply buried residue of the binding interface. It is positioned within a hydrophobic pocket such that addition of a hydroxyl group to its β-carbon is predicted from the structures to disrupt binding of the CAD to p300, illustrating the molecular mechanism by which hydroxylation by FIH-1 prevents HIF-dependent transcription.38 In support of these conclusions, fluorescence polarisation binding assays of the CAD with p300 demonstrate almost complete abrogation of binding when the CAD is hydroxylated.41
There is subtle variation in the regulation of HIF-1α–CAD compared with that of HIF-2α–CAD, with HIF-1α peptide substrates more efficiently hydroxylated by FIH than HIF-2α substrates in vitro.29 Although there is quite high conservation at the amino-acid level between the HIF-1α and HIF-2α–CADs, hHIF-1α has a conserved alanine at position 804, whereas HIF-2α has a valine in the equivalent position. As stated previously, this residue forms a hydrogen bond required for the correct orientation of the target asparagine into the active site of FIH-1. Mutational analyses that swapped the residues in this position (Ala804Val in HIF-1α and Val848Ala in HIF-2α) were sufficient to completely reverse the in vitro hydroxylation rates mediated by FIH-1 such that HIF-1α Ala804Val exhibited a Vmax similar to that of wild-type HIF-2α, and HIF-2α Val848Ala had an elevated Vmax much like that of wild-type HIF-1α.36
Localisation and Expression of FIH
FIH-1 protein expression has been detected at similar levels in all tissue culture cell lines investigated to date.34, 36 Immunohistochemical interrogation of an extensive range of human tissues revealed widespread and predominantly cytoplasmic staining of varying intensity for FIH-1 protein.42 During normoxia, FIH-1 is localised predominantly in the cytoplasm, and is available for modification and silencing of HIF-α. Treatment of cultured cells with hypoxia or hypoxia mimetics does not appear to alter the cytoplasmic localisation of FIH-1.34, 40, 43
Other FIH-1 Substrates and HIF Regulation
Until recently, knowledge of FIH-1 function was limited to its regulation of the HIF-α proteins. Recent studies have uncovered a new class of interacting proteins, with some being subsequently identified as genuine substrates for asparaginyl hydroxylation. All these proteins contain ankyrin-repeat domains (ARDs) and include IκBα; p105; Notch1, 2 and 3; and Ankyrin repeat and SOCS Box protein 4 (ASB4).44, 45, 46 Presently, the function of hydroxylation on these novel substrates remains unclear, with only subtle downstream effects, if any, being observed. However, it has been suggested that hydroxylation of ARD-containing proteins occurs in competition with HIF hydroxylation, and could potentially regulate HIF-mediated transcription by sequestering FIH-1, particularly in hypoxia.45
Regulation of FIH-1
As FIH-1 is a crucial regulator of the CAD, modulation of the activity of FIH-1 has important implications for transcriptional activity of HIF. In addition to regulation by intracellular oxygen levels, FIH-1 transcription and stability may also be regulated. FIH-1 has been shown to interact in vitro with the von Hippel–Lindau protein, which is an integral component of the ubiquitin ligase machinery responsible for oxygen-dependent degradation of HIF-1α, and histone deacetylases, although the purpose of this interaction is unclear.26 Recent reports link FIH-1 with another ubiquitin ligase Siah-1.47 While this research demonstrates an interaction between the two proteins that results in polyubiquitylation and degradation of FIH-1, the functional relevance remains ambiguous. Although the authors argue that it represents a hypoxia-dependent degradation mechanism for FIH-1 and thus a way of enhancing HIF transactivation under oxygen stress, there is no discernable increase in FIH-1 protein levels in hypoxia in this or previous studies.34, 47
A novel mechanism of regulation of FIH-1 transcription in renal carcinoma cells has been put forth following an earlier finding that FIH-1 message was increased with knockdown of protein kinase C-ζ (PKCζ), or overexpression of a dominant-negative form.48 Li and co-workers identified a cis-regulatory element in the FIH-1 promoter that binds phosphorylated CDP/Cut proteins and thus represses transcription. PKCζ is the kinase responsible for CDP/Cut phosphorylation, and phosphorylation by PKCζ was demonstrated to be important for DNA binding by CDP/Cut, and for FIH-1 repression.
As already mentioned, FIH-1 requires 2-OG, Fe2+, ascorbate and dioxygen for catalysis of hydroxylation. Regulation of all four HIF hydroxylases by the citric acid cycle intermediates fumarate, succinate and oxaloacetate was investigated, with the PHDs exhibiting negative regulation with all three.49, 50 FIH-1, however, was not inhibited by physiologically relevant levels of these compounds, and is thus unlikely to be regulated via metabolic pathway intermediates, possibly due to its more important role in other processes not directly related to cellular metabolism, including neural differentiation and vasculogenesis. Understanding of the regulation of FIH-1 expression and the consequences for HIF activity is limited and warrants further investigation.
Other Modifications Affecting HIF-Driven Transcription
In addition to asparagine hydroxylation, various posttranslation modifications have been reported to occur on the HIF-1α CAD, which can potentially modulate its activity. For example, phosphorylation of hHIF-1α at Thr796 by casein kinase II was first postulated by Gradin et al.51, and substitution of an aspartic acid residue at this position to mimic phosphorylation increased the interaction of the CAD with p300 within cells. It was subsequently shown that a peptide phosphorylated at this position could not be hydroxylated by FIH-1,52 and the aspartic acid mutation significantly decreased hydroxylation efficiency in vitro.53 Finally, in vitro interaction assays demonstrated that phosphorylation alone at this position did not enhance binding to p300,41 and rather indicated that phosphorylation at Thr796 inhibited hydroxylation by FIH-1, facilitating increased p300 binding and CAD activity.
Mitogen-activated protein kinase (MAPK) has been correlated with CAD activity as enhanced activation of MAPK increases HIF-α-mediated transactivation, and conversely inhibition of MAPK restricts transcription by disruption of the HIF-α-CAD/p300 interaction.54 Additionally, it has been reported that MAPK-mediated phosphorylation of serine residues 641 and 643 within the ID of HIF-1α reduces activity by preventing CRM1-dependent nuclear translocation of the subunit.55
S-nitrosylation of the thiol group of Cys800 of HIF-1α has been reported to increase transcriptional activity. This was postulated to occur via enhancement of the interaction between the CAD and CBP/p300, thus linking cellular redox status to recruitment of CBP to HIF.56 However, more recent quantitative interaction studies with purified proteins and peptides demonstrated a significant decrease in p300 binding upon Cys800 S-nitrosylation.41
SUMOylation of HIF-1α has also been described by several groups, however the reported outcomes of this modification are varied. For example, in a number of studies, SUMOylation of HIF-1α has been suggested to both increase HIF-1α stability and transcriptional activity,57, 58 but in others, it has been suggested to decrease activity and enhance VHL-mediated ubiquitination59, 60
Transcriptional Cofactors
Numerous factors in addition to CBP/p300 have been found to participate in HIF-mediated transcription. These can act ubiquitously or can discriminate between HIF-α isoforms or between specific target genes, and may also assist in tissue-specific gene expression. Examples include members of the p160 family of coactivators, such as TIF-2 and SRC-1, which both enhance HIF-1-mediated transactivation during hypoxia.20 HIF-1β is also responsible for recruitment of the reportedly essential coactivator thyroid hormone receptor/retinoblastoma-interacting protein-230 (TRIP230), although this does not require the characterised HIF-1β transactivation domain.22
HIF-1α and HIF-2α are closely related in domain architecture, DNA binding and activation mechanisms, however they exhibit non-redundancy as demonstrated by mouse knockout models. As yet, there is limited knowledge of the mechanisms that determine this non-redundancy, but differential regulation via protein recruitment and transactivation may play an important role. For example, HIF-2α but not HIF-1α binds the NF-kappaB essential modulator (NEMO), and this interaction assists in recruiting CBP/p300 and enhances HIF-2α-mediated transcription activity at normoxia.61 Also, the transcription factor Ets1 has been demonstrated to interact specifically with HIF-2α for the transcription of vascular endothelial growth factor (VEGF) receptor 2 , or Flk1.62
Examples of accessory transcription molecules that are required for or enhance transcription of specific target genes include hepatocyte nuclear factor 4 (HNF-4) and Smad3. HNF-4 is a liver- and kidney-specific transcription factor reported to be required for EPO transcription at hypoxia via binding to two adjacent repeats of the HNF-4 element in close proximity to the HRE present in the EPO enhancer.63 Mutation of the HNF-4 element, and thus loss of HNF-4 binding, prevented hypoxic induction of EPO. HNF4 has also been shown to interact directly with both HIF-1α and Arnt, and these interactions were deemed important for hypoxia-induced EPO transcription in the liver and kidney.64 HIF-1α was shown to interact with Smad3 in vivo, and to bind synergistically with HIF-1 to a Smad-binding site near the HRE in the VEGF promoter.65 As Smad3 was previously shown to interact with CBP/p300, a possible mechanism for the enhancement of VEGF transcription by Smad3 may be through stabilisation of the HIF-1α–CBP/p300 interaction on the DNA. Additionally, STAT3 was found to be important for maximal VEGF expression in complex with HIF-1 and CBP/p300 in prostate and pancreatic carcinoma.66 Conversely, the HIF proteins can also participate in transcriptional regulation of non-canonical pathways such as Myc and Notch signalling, where they are recruited to DNA via interaction with other transcription factors.67 Finally, Hsp90 has been shown in a number of studies to be required for HIF activity, although this appears to be predominantly through regulation of HIF stability as described previously.68
Conclusion
The regulation of HIF-mediated transcription has been the topic of intensive research in recent years, with the discovery and characterisation of the hydroxylation-mediated ‘hypoxic switch’ a major achievement among others. However, our understanding of this crucial signalling pathway remains incomplete. For example, the discovery of new interacting proteins and substrates for FIH-1 has important implications at the molecular and physiological level for HIF regulation and cross talk between the HIF and other pathways. Continued research in this field will not only enhance our understanding of this crucial pathway, but also contribute to our knowledge of the pathophysiology of major human diseases, and ultimately may facilitate the generation of novel therapeutic strategies.
Abbreviations
- 2-OG:
-
2-oxoglutarate
- ARD:
-
ankyrin-repeat domain
- Arnt1:
-
aryl hydrocarbon receptor nuclear translocator
- bHLH:
-
basic helix–loop–helix
- CAD:
-
C-terminal transactivation domain
- CBP:
-
Creb-binding protein
- CH1 domain:
-
cysteine/histidine-rich domain 1
- EPO:
-
erythropoietin
- FIH-1:
-
factor-inhibiting HIF-1α
- HBS:
-
HIF-binding site
- HIF:
-
hypoxia-inducible factor
- HRE:
-
hypoxia response elements
- Hsp90:
-
heat-shock protein 90
- IPAS:
-
inhibitory PAS domain protein
- NAD:
-
N-terminal transactivation domain
- ODDD:
-
oxygen-dependent degradation domain
- PAS:
-
Per-Arnt-Sim homology
- PHD:
-
prolyl hydroxylase domain
- SUMO:
-
small ubiquitin-like modifier
References
Wang GL, Jiang BH, Rue EA, Semenza GL . Hypoxia-inducible factor 1 is a basic-helix–loop–helix–PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92: 5510–5514.
Bracken CP, Whitelaw ML, Peet DJ . The hypoxia-inducible factors: key transcriptional regulators of hypoxic responses. Cell Mol Life Sci 2003; 60: 1376–1393.
Jiang BH, Semenza GL, Bauer C, Marti HH . Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 1996; 271: C1172–C1180.
Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA . Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 1998; 7: 205–213.
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001; 414: 550–554.
Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L . Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem 2002; 277: 32405–32408.
Heidbreder M, Frohlich F, Johren O, Dendorfer A, Qadri F, Dominiak P . Hypoxia rapidly activates HIF-3alpha mRNA expression. FASEB J 2003; 17: 1541–1543.
Huang LE, Arany Z, Livingston DM, Bunn HF . Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem 1996; 271: 32253–32259.
Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL . Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol 1998; 275: L818–L826.
Liu YV, Semenza GL . RACK1 vs HSP90: competition for HIF-1 alpha degradation vs stabilization. Cell Cycle 2007; 6: 656–659.
Chapman-Smith A, Whitelaw ML . Novel DNA binding by a basic helix–loop–helix protein. The role of the dioxin receptor PAS domain. J Biol Chem 2006; 281: 12535–12545.
Semenza GL, Wang GL . A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 1992; 12: 5447–5454.
Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, Hashimoto K et al. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J Biol Chem 2001; 276: 2292–2298.
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM . Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J Biol Chem 2006; 281: 15215–15226.
Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL . Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem 1997; 272: 19253–19260.
Pugh CW, O'Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ . Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem 1997; 272: 11205–11214.
Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM . The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res 2006; 66: 3688–3698.
Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 1996; 93: 12969–12973.
Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K et al. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 1999; 18: 1905–1914.
Carrero P, Okamoto K, Coumailleau P, O'Brien S, Tanaka H, Poellinger L . Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol 2000; 20: 402–415.
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML . Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002; 295: 858–861.
Beischlag TV, Taylor RT, Rose DW, Yoon D, Chen Y, Lee WH et al. Recruitment of thyroid hormone receptor/retinoblastoma-interacting protein 230 by the aryl hydrocarbon receptor nuclear translocator is required for the transcriptional response to both dioxin and hypoxia. J Biol Chem 2004; 279: 54620–54628.
Hu CJ, Sataur A, Wang L, Chen H, Simon MC . The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell 2007; 18: 4528–4542.
O'Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW . Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J Biol Chem 1999; 274: 2060–2071.
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK . FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 2002; 16: 1466–1471.
Mahon PC, Hirota K, Semenza GL . FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001; 15: 2675–2686.
Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 2002; 277: 26351–26355.
Hewitson KS, McNeill LA, Elkins JM, Schofield CJ . The role of iron and 2-oxoglutarate oxygenases in signalling. Biochem Soc Trans 2003; 31: 510–515.
Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J . Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem 2004; 279: 9899–9904.
Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J . Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem 2003; 278: 30772–30780; e-pub 2003 June 3.
Schofield CJ, Ratcliffe PJ . Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun 2005; 338: 617–626.
Fandrey J, Gorr TA, Gassmann M . Regulating cellular oxygen sensing by hydroxylation. Cardiovasc Res 2006; 71: 642–651.
Bell EL, Chandel NS . Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem 2007; 43: 17–27.
Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM et al. Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (HIF) in regulating HIF transcriptional target genes. J Biol Chem 2004; 279: 42719–42725.
Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ et al. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem J 2007; 401: 227–234.
Bracken CP, Fedele AO, Linke S, Balrak W, Lisy K, Whitelaw ML et al. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem 2006; 281: 22575–22585.
Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE . Structural basis for Hif-1 alpha /CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA 2002; 99: 5271–5276.
Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA 2002; 99: 5367–5372.
Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW et al. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem 2003; 278: 1802–1806.
Linke S, Stojkoski C, Kewley RJ, Booker GW, Whitelaw ML, Peet DJ . Substrate requirements of the oxygen-sensing asparaginyl hydroxylase factor-inhibiting hypoxia-inducible factor. J Biol Chem 2004; 279: 14391–14397.
Cho H, Ahn DR, Park H, Yang EG . Modulation of p300 binding by posttranslational modifications of the C-terminal activation domain of hypoxia-inducible factor-1alpha. FEBS Lett 2007; 581: 1542–1548.
Soilleux EJ, Turley H, Tian YM, Pugh CW, Gatter KC, Harris AL . Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 2005; 47: 602–610.
Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M et al. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci 2003; 116: 1319–1326.
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA 2006; 103: 14767–14772.
Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem 2007; 282: 24027–24038.
Ferguson III JE, Wu Y, Smith K, Charles P, Powers K, Wang H et al. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol 2007; 27: 6407–6419.
Fukuba H, Yamashita H, Nagano Y, Jin HG, Hiji M, Ohtsuki T et al. Siah-1 facilitates ubiquitination and degradation of factor inhibiting HIF-1alpha (FIH). Biochem Biophys Res Commun 2007; 353: 324–329.
Li J, Wang E, Dutta S, Lau JS, Jiang SW, Datta K et al. Protein kinase C-mediated modulation of FIH-1 expression by the homeodomain protein CDP/Cut/Cux. Mol Cell Biol 2007; 27: 7345–7353.
Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J . The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem 2006; 281: 28712–28720.
Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem 2007; 282: 3293–3301.
Gradin K, Takasaki C, Fujii-Kuriyama Y, Sogawa K . The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J Biol Chem 2002; 277: 23508–23514.
Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW et al. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J 2004; 383: 429–437.
Peet D, Linke S . Regulation of HIF: asparaginyl hydroxylation. Novartis Found Symp 2006; 272: 37–49, 50–53, 131–140.
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J . MAPK signaling upregulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem 2003; 278: 14013–14019.
Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 2006; 281: 33095–33106.
Yasinska IM, Sumbayev VV . S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett 2003; 549: 105–109.
Carbia-Nagashima A, Gerez J, Perez-Castro C, Paez-Pereda M, Silberstein S, Stalla GK et al. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 2007; 131: 309–323.
Bae SH, Jeong JW, Park JA, Kim SH, Bae MK, Choi SJ et al. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun 2004; 324: 394–400.
Berta MA, Mazure N, Hattab M, Pouyssegur J, Brahimi-Horn MC . SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem Biophys Res Commun 2007; 360: 646–652.
Cheng J, Kang X, Zhang S, Yeh ET . SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007; 131: 584–595.
Bracken CP, Whitelaw ML, Peet DJ . Activity of hypoxia-inducible factor 2alpha is regulated by association with the NF-kappaB essential modulator. J Biol Chem 2005; 280: 14240–14251.
Elvert G, Kappel A, Heidenreich R, Englmeier U, Lanz S, Acker T et al. Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1). J Biol Chem 2003; 278: 7520–7530.
Zhang W, Tsuchiya T, Yasukochi Y . Transitional change in interaction between HIF-1 and HNF-4 in response to hypoxia. J Hum Genet 1999; 44: 293–299.
Tsuchiya T, Kominato Y, Ueda M . Human hypoxic signal transduction through a signature motif in hepatocyte nuclear factor 4. J Biochem (Tokyo) 2002; 132: 37–44.
Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C . Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem 2001; 276: 38527–38535.
Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005; 24: 3110–3120.
Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 2005; 9: 617–628.
Katschinski DM, Le L, Schindler SG, Thomas T, Voss AK, Wenger RH . Interaction of the PAS B domain with HSP90 accelerates hypoxia-inducible factor-1alpha stabilization. Cell Physiol Biochem 2004; 14: 351–360.
Acknowledgements
We thank the members of the Peet laboratory for reviewing the manuscript; Anne Chapman-Smith for assistance with preparation of the figures and the Australian Research Council, National Health and Medical Research Council of Australia and the Cancer Council of South Australia for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by N Chandel
Rights and permissions
About this article
Cite this article
Lisy, K., Peet, D. Turn me on: regulating HIF transcriptional activity. Cell Death Differ 15, 642–649 (2008). https://doi.org/10.1038/sj.cdd.4402315
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402315
Keywords
This article is cited by
-
PHD1-3 oxygen sensors in vivo—lessons learned from gene deletions
Pflügers Archiv - European Journal of Physiology (2024)
-
Interplay of hypoxia-inducible factors and oxygen therapy in cardiovascular medicine
Nature Reviews Cardiology (2023)
-
m6A methylation regulates hypoxia-induced pancreatic cancer glycolytic metabolism through ALKBH5-HDAC4-HIF1α positive feedback loop
Oncogene (2023)
-
Role of Oxidative Stress in the Occurrence and Development of Cognitive Dysfunction in Patients with Obstructive Sleep Apnea Syndrome
Molecular Neurobiology (2023)
-
Worenine reverses the Warburg effect and inhibits colon cancer cell growth by negatively regulating HIF-1α
Cellular & Molecular Biology Letters (2021)
