
Abstract
So far, most monoclonal antibodies have been developed for treating cancer or immunological diseases. However, the global spread of infections such as West Nile and corona viruses, and the need to address the potential threat of bioterrorism, has boosted public interest in, and government support of, counter-measures for infectious diseases. The attractive features of monoclonal antibodies, such as high specificity and effective recruitment of the immune system, would seem to make them excellent candidates as anti-infective agents. Here, we analyse trends in the development and approval of anti-infective monoclonal antibodies, and discuss factors that influence their success.
Similar content being viewed by others

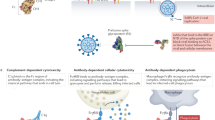

Main
Over the past decade, monoclonal antibodies (mAbs) have become a thriving class of therapeutics, with 18 having received regulatory approval, and more than 150 now in clinical development. The majority of such agents have been developed for treating cancer and immune disorders owing, at least in part, to the well-defined unmet medical needs and markets in these areas.
However, mAbs have also been investigated for their potential as anti-infective agents, although with limited success so far; of the 18 mAbs approved by the US FDA, only one is an anti-infective agent. Nevertheless, numerous candidates are undergoing preclinical and clinical study, and ample medical needs exist for targeted anti-infective treatments. With careful selection of indications and mAb characteristics (such as specificity, avidity and isotype), opportunities exist for the development of safe and efficacious anti-infective mAbs that would complement the arsenal of vaccines and anti-infective drugs. Some caution is advisable because, despite an increase in government funding for development, the markets for countermeasures to many pathogens have not yet been clearly defined.
As part of ongoing efforts to track trends in the development and approval of therapeutic mAbs1,2,3,4, Tufts Center for the Study of Drug Development collected data for commercially sponsored anti-infective mAbs at the preclinical and clinical stages of development (see Box 1 for inclusion criteria). Probabilities of success were calculated for products that entered clinical study in the 1980s and the 1990s. Vaccines and anti-infective drugs are potential market competitors, so we compared the anti-infective mAbs in clinical development with marketed and pipeline vaccines and anti-infective drugs and assessed overlap between target organisms.
Nascent technology: 1980s
The technology for the production of mAbs was developed in the late 1970s. Although more than 70 therapeutic mAbs entered clinical study in the 1980s, only 11 were anti-infective products. The concept of mAbs as 'magic bullets' capable of homing in on specific targets caught the imagination of scientists, businesspeople and the public, but in practice the idea was most commonly applied to cancer therapeutics. A 'war on cancer' had been declared in 1971, but efficacious oncology products were still very much in demand a decade later. Nevertheless, a number of companies explored the judicious use of anti-infective mAbs in areas in which medical need was as yet unmet by vaccines or anti-infective drugs — for example, cytomegalovirus, hepatitis B virus, human immunodeficiency virus (HIV) and human rhinovirus infections, and sepsis.
The original mAb production method — the fusion of mouse lymphocyte and myeloma cells — provided murine mAbs. These products were often immunogenic6, and so human mAbs were preferred as therapeutics. Of the eleven anti-infective mAbs in clinical study in the 1980s, seven (64%) were human, and only four (36%) were murine products. At the time, human mAbs were derived from human B-lymphocytes. As naturally infected patients were the source of these cells, only a limited number of pathogens could be targeted.
The early hybridoma methods lent themselves to preparation of immunoglobulin M (IgM) mAbs as well as the more versatile IgG products5. Of the eleven mAbs, four (36%) were IgM antibodies. However, IgM antibodies are composed of five monomers, with four protein chains comprising each monomer, and so have limited utility as therapeutics because of their large size. IgM antibodies are largely confined to the circulatory system, and so these products were studied in the 1980s primarily as treatments for bloodstream bacterial infections (that is, sepsis). In contrast, the majority (86%) of the IgG mAbs were studied as treatments for viral infections.
The overall approval success rate for anti-infective mAbs studied in the 1980s was zero. Of the eleven products that entered clinical study, five products (45%) progressed to Phase II, three (60%) progressed to Phase III and two (67%) advanced to FDA review, but neither application was approved. The two products reviewed by the FDA were edobacomab (XOMEN-E5; XOMA) and nebacumab (Centoxin; Centocor), both IgM antibodies studied as treatments for Gram-negative septicaemia. Additional efficacy testing was requested during the FDA review of edobacomab, but the product did not meet the endpoint (improvement of short-term survival) in an additional study7. Development of nebacumab was terminated due to excess mortality in sepsis patients who were treated but later diagnosed without Gram-negative bacteraemia8. Nebacumab was approved in a number of countries outside the US, and then voluntarily withdrawn from these markets.
Limited success: 1990s
Technological advances opened new opportunities in the 1990s when genetically engineered mAbs such as chimeric, humanized and bispecific products began entering the clinic in large numbers. Phage-display and transgenic mouse technologies made human mAbs readily available for development. More than 140 therapeutic mAbs entered clinical study during the 1990s, double the number studied in the 1980s. However, perhaps because of the high-profile product terminations of the previous decade, only 13 mAbs were studied as anti-infective agents.
Anti-infective mAb development in the 1990s was focused on the antiviral therapeutics, which comprised 77% of the total. Study of anti-HIV mAbs intensified: only one anti-HIV mAb was studied in the 1980s, whereas six of these products were in the clinic in the 1990s. The variety of the specificities was not large though: four (67%) mAbs targeted regions of HIV glycoprotein 120.
The overall approval success rate calculated for anti-infective mAbs studied in the 1990s was 10%; three mAbs remain at Phase II of clinical study. The rate increased slightly (to 11%) when the two murine products were removed from the analysis. Clinical phase-transition probabilities (the probability of moving from one clinical phase to the next) were 73%, 50% and 33% for transitions from Phase I to II, Phase II to III, and Phase III to US approval, respectively (Fig. 1). Only one anti-infective mAb studied in the 1990s, palivizumab (Synagis; MedImmune), was reviewed by the FDA; it was approved in June 1998 as a treatment for respiratory syncytial virus infection in high-risk paediatric patients.

*Review to approval transitions were 100% for all cohorts, so transition probabilities for Phase III to FDA review and FDA review to approval were combined.
We have previously published phase-transition probabilities for humanized mAbs stratified by time and therapeutic category4. Humanized mAbs comprise 50% of the US-approved mAbs and are the most abundant of the three main varieties of mAbs (chimeric, humanized and human) in clinical study. Phase-transition probabilities for anti-infective mAbs developed in the 1990s fell short of benchmarks set by humanized mAbs in clinical study during a similar period, as well as most of those for oncological and immunological diseases (Fig. 1). The problematic step for the anti-infective mAbs was successful completion of Phase III. Of the three non-murine anti-infective mAbs that entered Phase III, only one (palivizumab) was successful, whereas two were terminated in Phase III due to lack of efficacy.
Clinical monoclonal pipeline: 2000–2005
Industry focus on mAb products has intensified further in the 2000s. More than 130 therapeutic mAbs entered clinical study between January 2000 and June 2005. If this pace continues, the total for the decade could approach 240 products. However, as was the case in the two previous decades, anti-infective mAbs still comprise a small percentage of the total products in development to date. In the first 5.5 years of the 2000s, 22 mAb treatments for infectious diseases entered clinical study. Most are at early stages of development, with 11 (50%) at Phase I and nine (41%) at Phase II. Only two (9%) have entered Phase III studies (Table 1). The average length of the clinical development phase for FDA-approved mAbs was 6.5 years, so it is not surprising that these products are still undergoing evaluation.
Success rates cannot be calculated for anti-infective mAbs comprising the current 2000s cohort because all of the agents are still under clinical study. Predictions based on success rates for the 1990s cohort suggest that two or three of the products might be approved. However, due to the small size of the 1990s cohort, this result must be considered an estimate. In addition, a number of factors might improve success rates for the 2000s cohort, which includes more human mAbs and a number of mAbs with fast-track designation.
Genetically engineered human mAbs comprise the majority (59%) of the 22 anti-infective products and might have higher probabilities of success compared with chimeric or humanized products. Accurate assessment of success rates is not yet possible because few human mAbs in any therapeutic category have completed the development process. Only one is FDA-approved so far: adalimumab (Humira; Abbott), which received approval on 31 December 2002, as a treatment for rheumatoid arthritis.
Four mAbs in the current 2000s cohort have FDA fast-track designations as treatments for bacterial (Bacillus anthracis, Staphylococcus) and viral infections (hepatitis C, HIV). Available only since 1997, fast-track designation is intended to facilitate the development of medicinal products that might be improvements over the existing therapies available for patients with serious or life-threatening diseases. Companies sponsoring fast-track products are eligible for frequent and timely interactions with FDA. Fast-track product applications are eligible for 'rolling review', in which sections can be filed over time. Although these benefits will obviously not affect the overall safety or efficacy of the products, they might improve the quality of the clinical development programme and marketing application, increase the speed of the FDA review and decrease the likelihood of a termination for commercial reasons.
Potential clinical candidates
We examined company preclinical pipelines to assess the number of anti-infective mAbs that might enter the clinic in the remainder of the decade. Products and company programmes were assigned to three categories: the 'preclinical' category comprising discrete, clearly defined mAbs that had been tested in animals; the 'early research' category comprising single mAbs or groups of several mAbs that had in vitro data (for example, target organism neutralization data); and the 'screening' category comprising mAb library generation and screening programmes.
Preclinical mAb products and programmes sponsored solely by government and academic institutions were excluded. Because discoveries by these institutions might be licensed for commercialization in the future, our results have likely undercounted the total possible mAb candidates by an unknown amount.
We found that the preclinical group comprised only eleven products under investigation by eight companies. Interestingly, the selection of targets seemed to be strongly influenced by the Category A, B and C priority pathogens list used by US Centers for Disease Control and Prevention and National Institutes of Health (NIH)9. Category A agents are the highest priority and include pathogens that are easily disseminated or transmitted person-to-person. Of the preclinical products, eight (73%) were targeted to a Category A, B or C pathogen (Table 2). Nearly half the products were potential treatments for either anthrax (Category A; three mAbs) or West Nile virus (Category B; two mAbs) infection. By contrast, only six (24%) of the mAbs currently in clinical studies and none of the now discontinued mAbs were directed towards pathogens on the priority pathogens list.
The early research and screening groups contain the most speculative products and programmes. A total of 21 potential products from 17 companies were identified in early research. Though not present in the preclinical group, mAb therapeutics for HIV and hepatitis B and C infection reappear in the early research group. Only four mAbs for priority pathogens were included — one each for anthrax, rabies, severe acute respiratory syndrome (SARS) and variola infection. The screening group comprised anti-infective mAb programmes from 15 companies. Screening programmes for mAbs targeted to HIV, hepatitis A, B and C, as well as four priority pathogens — Clostridium botulinum, Ebola virus, SARS-associated corona virus and variola virus — were identified. Targets for five programmes were not disclosed. It remains to be seen whether any of these programmes will generate clinical candidates.
Competitive pressure
Companies developing anti-infective mAbs are jockeying for position in the market with two other groups of products: vaccines and anti-infective drugs. The presence of competitive products could be one explanation for the dearth of anti-infective mAbs in development. We examined marketed and pipeline vaccines and anti-infective drugs to assess whether these products might provide competition to mAbs in development. We focused on products targeting hepatitis C and HIV viral infections, and B. anthracis, pathogenic Escherichia coli, and Staphylococcus bacterial infections because most (76%) of anti-infective mAbs in the clinic (Table 2) are treatments for these five pathogens.
Although in theory prevention of infection would be desirable compared with treatment, a vaccine is currently marketed in the US for only one of the relevant pathogens — anthrax. The anthrax vaccine is not convenient — six doses must be administered over an 18-month interval — and is recommended only for persons thought to be at risk of exposure.
Vaccines are in development for all of the five target organisms, though all are in early stage clinical studies. Eight vaccines are in Phase I and four vaccines are in Phase II studies for hepatitis C, B. anthracis, pathogenic E. coli or Staphylococcus infections. At least 16 HIV vaccines are currently in early clinical studies sponsored at least in part by companies. Non-profit institutions and government agencies worldwide have been involved in the development of many HIV vaccines10 and the extent of commercial involvement in the development of some is unclear. As is well known, none of the HIV vaccines have as yet shown a great deal of promise.
Anti-infective drugs are marketed as treatments for all five pathogens. However, many drugs for the viral pathogens are not exceptionally effective because they inhibit but do not cure infection, and some cause serious side effects. The antibacterial drug ciprofloxacin (Bayer) is efficacious for mild cases of cutaneous anthrax, though it is not as effective against the inhaled pathogen. Infections with pathogenic E. coli can be managed with supportive care, though development of haemolytic uremic syndrome in infected paediatric patients remains a serious problem. A number of drugs are effective against Staphylococcus, but some strains are now resistant to many of these drugs.
A number of anti-infective drugs are in early stage clinical studies as treatments for hepatitis C and HIV. Two are currently in Phase II studies: viramidine (Valeant Pharmaceuticals) for hepatitis C and maraviroc (Pfizer) for HIV. Very few products are in studies as treatments for the three bacterial pathogens; none are in late-stage development. Pathogenic E. coli infections pose an especially difficult challenge for anti-infective drug development. The non-pathogenic strains are beneficial intestinal flora and anti-infective drugs are usually not selective enough to differentiate one from the other variety.
Cautious optimism: 2006 and beyond
As technology advances in the 2000s, companies are increasingly exploiting the benefits of mAbs (for example, high specificity and effective recruitment of the immune system), while reducing obstacles to their development (for example, anti-mAb immune reactions and complex production). In general, the unique functionalities and higher probabilities of approval success of mAbs compared with small molecules make them attractive for diversification of company pipelines. However, only a small number of anti-infective mAbs are currently under clinical study and few seem ready to enter the clinic.
Challenges specific to the development of mAbs as anti-infective agents have clearly caused companies to proceed with caution in this area. Treatment with mAbs is passive immunization, but unlike vaccination, anti-infective mAbs can provide only short-term prophylaxis. The parenteral routes of administration for mAbs are decidedly less convenient than the oral administration of many anti-infective drugs. Production of mAbs is complicated and costly compared with that of many vaccines and drugs. So far, the most optimistic overall approval success rate we calculated for anti-infective mAbs (11%) is lower than that for anti-infective drugs (16%) produced by major pharmaceutical firms11, though it must be noted that some of the approved anti-infective drugs are derivative versions of previously successful products, whereas the approved anti-infective mAb is novel. As a consequence of these factors, competition from vaccines and anti-infective drugs has focused mAb development in niche applications in which medical need exists for new treatments.
Opportunities for successful anti-infective mAb development do exist in these niche applications, however, and returns are not necessarily small. For example, the FDA-approved mAb treatment for respiratory syncytial virus in high-risk infants, palivizumab, had global sales of nearly a billion dollars in 2004. In addition, the landscape of anti-infectives development in the US has changed due to recent legislation, increased emphasis by the National Institutes of Health on priority pathogens, and changes to FDA regulations. Specifically, numerous opportunities are currently available for development of anti-infective mAbs as bioterrorism countermeasures.
The need for safe and effective anti-infective agents has taken on an added urgency since September 2001. Recent US legislation has made a substantial amount of money available. Approved 21 July 2004, the Project BioShield Act amended the Public Health Service Act specifically “to provide protections and countermeasures against chemical, radiological, or nuclear agents that may be used in a terrorist attack against the United States by giving the National Institutes of Health (NIH) contracting flexibility, infrastructure improvements, and expediting the scientific peer review process, and streamlining the Food and Drug Administration approval process of countermeasures”12. Authorization for appropriation of up to US$5.6 billion for fiscal years 2004 through 2013 was given to achieve the desired goals.
Project BioShield provides financial incentives (for example, government purchase contracts) to companies that invest in the development of bioterrorism countermeasures. However, it must be noted that so far few companies have been enticed by the opportunities, due in part to the lack of a clearly defined market for the resulting products13. The deficiencies of Project BioShield have been recognized and various new provisions and additional inducements are being considered for a revised version of the legislation14. The proposed Project BioShield II Act was under discussion by the US Senate Committee on Health, Education, Labor, and Pensions as of July 2005.
Another source of funding for the development of anti-infective agents is available through NIH 'challenge grants'. The programme specifically supports development of product candidates such as therapeutics and vaccines against Category A, B and C priority pathogens. Various phases of development can be funded through challenge grants, including early validation, production and preclinical and early clinical studies, though not all phases are covered in every request for applications issued by NIH.
Development of anti-infective agents can also be facilitated through use of FDA regulations colloquially referred to as the 'animal rule'. This rule was proposed in 1999 and published in final form in 200215. The animal rule states that FDA approval may be based on evidence of effectiveness from adequate and well-controlled studies in animals. However, safety in humans must also be established and results of animal studies must establish that the product is reasonably likely to produce clinical benefit in humans. Approvals under the rule therefore absolutely require the existence of appropriate animal models for the infectious diseases.
The animal rule regulations specify the evidence that is needed to demonstrate the effectiveness of new drugs and biological drug products when human efficacy studies are not ethical or feasible. These conditions exist for a number of anti-infective bioterrorism countermeasures because it is usually unethical to induce or encourage infection in human subjects as a means to study product effectiveness, and rigorous efficacy studies are not feasible in cases of uncommon natural infections, such as anthrax infection. A number of strings come attached to an approval under the rule — post-marketing studies must be done when such studies are ethical and feasible, the FDA may restrict distribution to ensure safe use, and patient labelling must explain that the approval was based on efficacy studies conducted in animals alone.
Anti-infective mAb development is a difficult endeavour, but the prevailing conditions have opened up new opportunities. Traditional approaches such as use of vaccines and anti-infective drugs have failed to control the spread of any number of infectious agents, so the need for novel therapeutics still exists. A greater appreciation for the ease of both intentional and unintentional spread of infectious agents has caused the government to make additional funding available for the development of anti-infective products and to modify regulations for their approval. Companies now need to capitalize on these opportunities to meet the need for new anti-infective mAbs, though some caution is advisable in cases in which the markets for these products are not well defined.
References
Reichert, J. M. Monoclonal antibodies in the clinic. Nature Biotechnol. 19, 819–822 (2001).
Reichert, J. M. Therapeutic monoclonal antibodies: trends in development and approval in the US. Curr. Opin. Mol. Ther. 4, 110–118 (2002).
Reichert, J. & Pavlou, A. Monoclonal antibodies market. Nature Rev. Drug Discov. 3, 383–384 (2004).
Reichert, J. M., Rosensweig, C. J., Faden, L. B. & Dewitz, M. C. Monoclonal antibody successes in the clinic. Nature Biotechnol. 23, 1073–1078 (2005).
Hector, R. F., Collins, M. S. & Pennington, J. E. Treatment of experimental Pseudomonas aeruginosa pneumonia with a human IgM monoclonal antibody. J. Infect. Dis. 160, 483–489 (1989).
Khazaeli, M. B., Conry, R. M. & LoBuglio, A. F. Human immune response to monoclonal antibodies. J. Immunother. 15, 42–52 (1994).
Angus, D. C. et al. E5 murine monoclonal antiendotoxin antibody in Gram-negative sepsis: a randomized controlled trial. JAMA 283, 1723–1730 (2000).
Inglis, T. J. J. et al. Monoclonal antiendotoxin agent HA-1A (Centoxin). Lancet 341, 303 (1993).
National Institutes of Health web site [online], <http://www2.niaid.nih.gov/biodefense/PDF/cat.pdf> (2005).
Garber, D. A., Silvestri, G. & Feinberg, M. B. Prospects for an AIDS vaccine: three big questions, no easy answers. Lancet Infect. Dis. 4, 397–413 (2004).
Kola, I. & Landis, J. Can the pharmaceutical industry reduce attrition rates? Nature Rev. Drug Discov. 3, 711–715 (2004).
Project BioShield Act of 2004. US Public Law 108–276 (2004 July 21); 42 USC 247d-6a; 1 18 Stat. 835.
Usdin, S. Bioshield hammered. BioCentury 13(32), A14 (2005).
Project BioShield II Act of 2005. S.975 (2005 April 29).
Federal Register. New drug and biological drug products; evidence needed to demonstrate effectiveness of new drugs when human efficacy studies are not ethical or feasible. 67, 37988–37998 (2002).
Acknowledgements
The authors gratefully acknowledge the assistance of the companies that provided survey data. We also thank G. Laroia for reviewing the manuscript and for providing helpful comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
OMIM
FURTHER INFORMATION
Rights and permissions
About this article
Cite this article
Reichert, J., Dewitz, M. Anti-infective monoclonal antibodies: perils and promise of development. Nat Rev Drug Discov 5, 191–195 (2006). https://doi.org/10.1038/nrd1987
Issue Date:
DOI: https://doi.org/10.1038/nrd1987
This article is cited by
-
The present state of the art in expression, production and characterization of monoclonal antibodies
Molecular Diversity (2016)
-
The safety and side effects of monoclonal antibodies
Nature Reviews Drug Discovery (2010)
-
Development trends for human monoclonal antibody therapeutics
Nature Reviews Drug Discovery (2010)
-
Preclinical safety testing of monoclonal antibodies: the significance of species relevance
Nature Reviews Drug Discovery (2007)
