
Abstract
Homozygous transgenic knockout mice without μ-opioid receptors lack morphine-induced antinociception, locomotion, tolerance, physical dependence, and reward. μ receptors thus appear to play central roles in these morphine actions. Different levels of μ receptor expression are found in different humans and in different animal strains. In vitro studies indicate that some morphine responses persist after inactivation of as many as 90% of the initial μ receptor complement, while others are attenuated after inactivating many fewer receptors. Varying levels of μ receptor reserve could thus exist in different μ-expressing neuronal populations in vivo. Heterozygous μ receptor knockout mice express half of wild-type μ receptor levels. Tests of morphine actions in these mice reveal evidence for differing μ receptor reserves in brain circuits that mediate distinct opiate effects. Heterozygotes display attenuated locomotion, reduced morphine self-administration, intact tolerance, rightward shifts in morphine lethality dose/effect relationships, and variable effects on place preference compared to wild-type mice. They demonstrate full physical dependence, as measured by naloxone-precipitated abstinence following five days of morphine administration. Neuroadaptive changes in sites other than μ receptors could be involved in some of these results. Nevertheless, these data document substantial influences that individual differences in levels of μ receptor expression could exert on distinct opiate drug effects. They support the idea that functional μ receptor reserve differs among the diverse neuronal populations that mediate distinct properties of opiate drugs.
Similar content being viewed by others

Main
The μ-opioid receptor (μ OR) is a seven-transmembrane domain, G protein-linked receptor that is a key member (Wang et al. 1993), along with the enkephalin-preferring δ (Kieffer et al. 1992) and dynorphin-preferring κ receptors (Minami et al. 1993), of the constellation of brain receptors that recognize morphine and other opiate drugs (Uhl et al. 1994). Pharmacologic approaches have suggested that the μ receptor, for which morphine has the highest affinity (Pasternak 1993), also serves as a principal site for morphine actions in inducing behavioral reward (Di Chiara and North 1992; Wise 1996), locomotion (Stevens et al. 1986), analgesia (Porreca and Burks 1993), tolerance (Aghajanian 1978), and physical dependence (Johnson and Fleming 1989). Neurons expressing μ receptors are found at many different levels of the neuraxis (Moriwaki et al. 1996). Many of these localizations can be correlated with some of the multiple actions of opiate drugs (Behbehani 1995).
Recent successes in developing transgenic knockout mice with μ receptor gene deletions (Matthes et al. 1996; Sora et al. 1997b; Tian et al. 1997; Loh et al. 1998) allow studies of morphine effects in animals that possess two, one, or no copies of the μ receptor gene. Heterozygous mice express half-normal levels of μ receptor expression in Scatchard analyses of [3H][D-Ala2, N-MePhe4, Gly5-ol] enkephalin (DAMGO) binding to whole brain and in immunohistochemical analyses of μ receptor immunoreactivity in spinal cord and several other examined brain regions that included sections through striatum and locus coeruleus (Sora et al. 1997b; I.S., R. Revay, and G.R.U., unpublished data). Homozygous knockout mice display no detectable μ receptor binding or immunoreactivity, although they contain near-normal complements of δ and κ receptors and of the mRNAs encoding these receptors, preproenkephalin, preprodynorphin, and preproopiomelanocortin (Matthes et al. 1996; Sora et al. 1997b). [35S]guanosine-5′-O-(3-thio)triphosphate (GTPγS) binding stimulated by endomorphin-1, endomorphin-2 or the synthetic selective μ-opioid receptor agonist DAMGO is reduced by approximately half in whole brain, brainstem or spinal cord of heterozygous mice and virtually eliminated in homozygous knockout mice (Narita et al. 1999; Mizoguchi et al. 1999). Homozygous μ receptor knockout mice are virtually devoid of the robust morphine-induced analgesia, physical dependence and reward that can be found in wild-type mice (Matthes et al. 1996; Sora et al. 1997b). This data supports near total μ-receptor dependence of these processes. However, it provides no information about influences of partial μ receptor depletions.
“Receptor reserve” is a concept that arose from inactivation experiments that examined the effects of eliminating various fractions of the available receptor pool. If, after inactivating some receptors, concentration-response curves display full efficacy with no depression of the maximal responses, receptor reserve is present (Chavkin and Goldstein 1984; Adams et al. 1990; Zernig et al. 1995). This phenomenon is also cited as evidence for “spare receptors” (Furchgott 1967; Ruffolo 1982; Kenakin 1984).
We now report studies on morphine responses in tests of reward, locomotion, lethality, tolerance, and physical dependence in mice expressing normal, half-normal, and absent μ receptor complements. We compare these data to work on analgesic responses obtained in the same lines of knockout mice (Sora et al. 1999, 1997a,b; Kitanaka et al. 1998; Fuchs et al. 1999; Qiu et al. 2000; Hosohata et al. 2000). The data should be interpreted in light of cautions about effects of adaptive mechanisms in these mice. Nevertheless, they provide a basis for identifying different relationships between μ receptor densities and acute or chronic morphine effects in some of the multiple neuronal systems whose functions are modulated by opiate drugs.
MATERIALS AND METHODS
Animals
Heterozygote-heterozygote matings of μ-opioid receptor knockout mice produced most wild-type, heterozygous and homozygous knockout animals on F2 and F3 C57/129 backgrounds that were identified by genotyping using Southern analyses as described (Sora et al. 1997b). For some experiments seeking influences of genetic background, progeny of eight-generation C57BL/6 backcross μ-opioid receptor congenic knockout mice, termed MUBX mice, were used. MUBX mice were derived by eight generations of successive backcrosses of heterozygous male knockout mice with C57BL/6 females (Jackson Labs). Subsequently, MUBX mice for pharmacological studies were generated by heterozygote crosses. Mice were housed at 24°C/50% relative humidity using a 12/12 hr light/dark cycle with lights on at 7:00 A.M. and off at 7:00 P.M. and ad libitum access to food and water under American Association for Laboratory Animal Care guidelines, as described (Sora et al. 1997b). Mice tested for each experiment in the morning, and an additional afternoon session added for place preference conditioning. Knockouts were compared to littermate controls to maintain identical average genetic backgrounds.
Locomotor Activity
Locomotor activity was assessed as total distance traveled. Distance was calculated from measurement of the number of beam breaks by mice placed individually in 46 × 25 × 19 cm clear plastic cages, to which the mice had not been previously exposed, in Optovarimex activity monitors (Columbus Instruments, Columbus, OH) under dim light, sound-attenuated conditions. Distance traveled was monitored for 1 hr, mice were injected with 10 mg/kg morphine hydrochloride, and distance traveled was monitored for an additional 3 hr (Miner and Marley 1995).
Conditioned Place Preference
Reward was assessed by conditioned place preference testing using a two compartment Plexiglas chamber (Miner et al. 1995). One compartment (18 × 18 × 18 cm) had a wire mesh floor (1.3 cm grids) mounted over Plexiglas. The other compartment (18 × 18× 18 cm) had corncob bedding on a smooth Plexiglas floor. An 18 × 5 × 1.3 cm platform was flush with the wall dividing two sides, which were separated by a removable Plexiglas wall. For pre- and post-conditioning test sessions, a 5-cm opening in the center wall allowed access to both compartments. During the conditioning sessions the opening was eliminated to restrict animals to a single compartment. Locomotion and time spent in each compartment was recorded using an Optivarimax animal activity monitoring apparatus (Columbus Instruments, Columbus, OH).
Conditioned place preferences were assessed by determination of compartment preference in three phases, as previously described (Suzuki et al. 1993). Initial preference, usually for the bedding floored compartment, was determined as the side in which a mouse spent more than 600 sec out of a 20-min trial. Conditioning was conducted over a 4-day period in which morphine was administered to the animal when it was confined for 30 min in the initially-nonpreferred compartment and saline administered to the animal when restricted for 30 min to the initially-preferred compartment. Animals received one conditioning session per day, counterbalanced between saline and morphine for a total of two saline pairings and two morphine pairings. Some mice received pretreatments with naloxone (10 mg/kg, s.c.) 10 min. prior to saline or morphine conditioning doses. Conditioned place preference assessments followed the last conditioning session and the last injection by 24 hours. Time spent in the drug-paired compartment was compared to the pre-conditioning values obtained on initial assessments. Dose effect analyses used s.c. morphine sulfate doses of 0, 1, 5, and 10 mg/kg (C57/129 mixed background) and 10 mg/kg (C57BL/6 congenic background MUBX).
Operant Morphine-Reinforced Behavior
Apparatus
Operant chambers used balanced rocker arm levers that broke infrared photo beams when 0.5 g of force was applied, solenoid systems adapted from Beardsley and Meisch (1981) that delivered 5 μl of water, microliter syringe pumps (Harvard 22, South Natick, MA) and stimulus lights controlled by integrated Coulborn environmental control systems with Med Associates interfaces, as previously described (Elmer et al. 1995a). PC-based system control, data acquisition and data storage used Med Associates software (St. Albans, VT).
Water Training and Surgery
To assess operant morphine self-administration behavior, mice were first trained on a modified progressive ratio schedule for water reinforcement. These data were used to confirm the ability of these mice to respond for a reinforcer and allowed assessments of differences in their abilities to respond to changing response criteria. Mice were water deprived for 24 hr, placed in the operant chamber, allowed access to 50 reinforcements of given 5–10 μl of water and a stimulus lights above a spout on an fixed ratio 1 (FR1) schedule, allowed access on a higher fixed ratio requirement (FRX + 1) for 50 trials, and so on. Subjects were run 24 hr/day for four days with free access to food. Mice were then anesthetized using ketamine (80 mg/kg, i.p.) and xylazine (16 mg/kg, i.p.). A 0.012″ i.d. silastic catheter was implanted in the right jugular vein, its tip advanced to the atrium, passed subcutaneously so that it exited in the midscapular region, and connected to a tether/swivel system (Instech, Plymouth Meeting, PA) mounted to the skull of the mouse with dental cement (Geristore, DenMat, CA).
Morphine Self-administration Behavior: FR4 Dose-Effect Curve
Some catheterized mice were tested on FR4 schedules of reinforcement for 1.0, 0.3, 0.1, and then 0 mg/kg/inj morphine to determine the presence or absence of morphine-reinforced behaviors, as well as morphine dose-effect functions. One to three days after implantation, mice were placed in operant chambers and allowed access to 1.0, 0.3, 0.1, or 0 mg/kg/injection morphine for 5, 3, 3, and 8 days respectively. Seven-eight mice of each genotype were run on FR4 schedules. Completion of each four responses illuminated an overhead light and a stimulus light above the spout and triggered a 5–8 μl morphine injection over 15 sec, followed by a 30 sec time-out period indicated by turning off the overhead and stimulus lights. After each time out, a stimulus light illuminated the lever and signaled morphine availability. Mice were run 24 hr/day with free access to food and water on a12 hr light/dark cycle.
Morphine Self-administration Behavior: Progressive Ratio (PR) Performance
Some catheterized mice were begun on a FR4 schedule and than changed to a progressive ratio schedule of morphine reinforcement using 1.0 mg/kg morphine per injection. These data were used to seek differences in morphine's efficacy in maintaining self-administration behavior. In this group of 68 mice of each genotype, mice were given access to 1.0 mg/kg morphine per injection for seven days on an FR4 schedule of reinforcement as described above, then placed on a progressive ratio schedule. Completion of each ratio resulted in an increase in the ratio required to obtain the next reinforcement. A stimulus light illuminating the lever signaled morphine availability The ratio requirements were 1, 3, 5, 7, 9, 12, 15, 18, 23, 8, 33, 41, 49, 57, 70, 83, 96, 117, 138, 156, 200, 225, 275, 300, 325, 350, 375, 425, 475, 525, and 600 (Roberts and Bennett 1993). Animals remained in the operant chamber for the duration of the experiments, where they were run 12 hr/day (20:00–08:00 hr) with free access to food and water and maintained on a 12-hr light dark cycle.
Morphine Antinociceptive Tolerance Testing
For hot-plate antinociception testing, mice were placed on a 55°C hot-plate and latency to hind-paw lick or jumping recorded with a 30 sec cutoff time in a standard hotplate paradigm (Dewey et al. 1969; Sora et al. 1997b). For tail-flick testing, mice were loosely wrapped in an adsorbent towel and their tails immersed approximately 2 cm into water heated to 53°C as previously described (Dewey et al. 1969; Sora et al. 1997b). The time after immersion at which the tail-flick response was recorded, with a cutoff time of 15 sec.
An ascending-dose cumulative dose-effect regimen was used, as described and as previously used with these and other transgenic mice (Elmer et al. 1995b). Morphine effects were assessed in naïve wild-type mice injected s.c. at 20-min intervals with an ascending cumulative dosing regimen that produced total drug doses of 3, 10, 30, and 56 mg/kg morphine. Higher cumulative doses were employed for tolerant and for heterozygote mice. Hot-plate and tail-flick responses were tested 20 min after each subcutaneous injection, and the next dose administered (Sora et al. 1997b). To induce tolerance and physical dependence, morphine was administered s.c. daily at 9 a.m. and 6 p.m. for 3, 5, or 7 days with doses ascending from 15 mg/kg by 5 mg/kg increments. Tolerance to morphine-induced antinociception was assessed 18 hr after the final morphine pretreatment by assessing the influences of acute s.c. injections with saline and then an ascending cumulative dosing regimen producing total drug doses of 10, 30, 56, and 100 mg/kg morphine using injections spaced 20 min apart. Hot-plate and tail-flick responses were tested 20 min after each subcutaneous injection, and the next dose then administered.
Physical Dependence
Morphine was administered s.c. daily at 9 a.m. and 6 p.m. for 3, 5, or 7 days, using a dose regimen ascending from 15 mg/kg by 5 mg/kg increments. Withdrawal was precipitated by administration of naloxone (1 mg/kg, s.c.) 2 hr after antinociceptive tolerance testing undertaken after cumulative dosing with 10, 30, 56, or 100 mg/kg morphine. Mice were placed into a novel Plexiglas chamber and naloxone-precipitated jumps counted over 15 min intervals by a blinded observer as described (Funada et al. 1994).
Morphine Lethality
Mice were observed for two hours following a single subcutaneous dose of 400, 500, 600, 650, 750, or 1000 mg/kg morphine sulfate. The latency to lethality was measured for each mouse that died, and the percent mortality for each genotype calculated. Three or four doses of morphine with 7–20 mice per dose in each genotype were used for each LD50 determination.
Drugs
All drugs were dissolved in saline and administered s.c. in a volume of 1.0 ml/kg. Naloxone (chloride) was purchased from Research Biochemical International, Inc (Natick, MA). Morphine (sulfate) was supplied by NIDA's Drug Supply Program.
Statistical Analyses
“Percent conditioned place preference” scores represent 100 × the ratio between the time spent in the environment paired with morphine in pre- and post-conditioning test sessions divided by the time spent in non-preferred environment before the training sessions: 100 × {[(time spent in post-conditioning) − (time spent in pre-conditioning)]/time spent in pre-conditioning]}. Self-administration: Genotype-dependent differences in water-reinforced behavior were analyzed using a two way (genotype × day) repeated-measures analysis of variance (ANOVA) with the number of reinforcements obtained across days as the dependent variable. Differences in fixed-ratio performance were analyzed using a two-way (genotype × dose) repeated-measures ANOVA using the number of lever presses and drug-intake as dependent variables.
Differences in progressive-ratio performance were analyzed using a two-way repeated-measures ANOVA (genotype × reinforcement schedule) with the number of reinforcements as the dependent variable. Percent maximal antinociception represents 100 × the ratio between the increased latency to paw lick in the morphine test condition over baseline values and the difference between the cutoff time and the baseline latency determined for each mouse: 100 × {[(latency to paw lick or tail flick after morphine) - (latency to paw lick or tail flick at baseline)]/[(cutoff time) - (baseline latency)]}. The cutoff time was 30 sec in hot-plate and 15 sec in tail-flick tests. ED50 values were calculated by linear regression of the ascending portions of dose effect curves. The relative potencies (RPs) were determined by dividing individual pretreatment ED50 values by post-treatment ED50 values. Most statistical comparisons were made using the Statistical Package for Social Science (SPSS Inc., Chicago, IL). Antinociceptive test data and the time intervals between morphine injection and lethality were analyzed by ANOVA following by Scheffe post-hoc analyses. Data for conditioned place preferences was analyzed using parametric and by nonparametric Kruskal-Wallis One-way ANOVAs followed by Mann-Whitney-Wilcoxon rank sum testing. Naloxone-induced jumping was analyzed by Mann-Whitney-Wilcoxon rank sum testing. Data are presented as a mean ± SEM in each of the experimental groups.
RESULTS
Morphine-Induced Locomotion
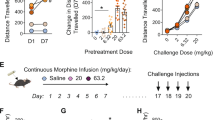
Homozygous and heterozygous μ knockout mice displayed exploratory behavior similar to wild-type mice during a one hour habituation period (Figure 1A ) (n = 6, 10, and 8, respectively). Morphine (10 mg/kg, s.c.) increased locomotion in well-habituated wild-type mice. However, homozygous μ knockout mice displayed no significant morphine-induced increase in locomotion when studied 1, 2, or 3 hr following morphine (Figure 1B ; ANOVA followed by Scheffe post-hoc analysis: F[2,21] = 5.6, p < .05, F[2,21] = 6.0, p < .05 and F[2, 21] = 3.6, p < .05 compared to wild-type controls, respectively). Morphine-induced locomotor activity in heterozygous mice was half that induced in wild-type control mice. Morphine-induced locomotion was thus gene-dose dependent. Interestingly, peak locomotor activity levels appeared to persist longer in wild-type than in heterozygous knockout mice (Figure 1B).

Spontaneous locomotion during habituation to a novel environment in wild-type, heterozygous, and homozygous μ-opioid receptor knockout mice. Locomotor activities of wild-type (+/+, open circles), heterozygous (+/−, closed circles), and homozygous (−/−, closed rectangles) μ-opioid receptor knockout mice (C57/129 background) were recorded for one hour in an activity monitor cage to which the mice had not been previously exposed. Values reflect distances traveled over 10-minute periods, in meters. No difference among genotypes reached statistical significance.

Attenuated morphine-induced locomotion in μ-opioid receptor knockout mice. Morphine (10 mg/kg, s.c.) injection increased locomotion in wild-type mice. The homozygous μ-opioid receptor knockout mice (C57/129 background) did not show any significant morphine-induced increase in locomotion when compared to wild-type control mice after 1, 2, and 3 hr morphine treatment. The heterozygous mice displayed significantly reduced morphine-induced locomotion when compared to wild-type control mice studied 2 hr after morphine administration (bar heights reflect distance traveled over a one hour period in meters; *p < .05 vs. wild-type group). These values can be compared to those of the end of the habituation period described in Figure 1A, basal level of locomotion without morphine, with attention to differences in scales.
Morphine-Induced Reward
Conditioned Place Preference
Mixed C57/129 wild-type mice displaying full μ receptor expression showed morphine conditioning (Figure 2A ) (overall Kruskal-Wallis one-way ANOVA by morphine dose, n = 44, χ2 = 9.9, p < .05) that was significant at 10 mg/kg (p < .05 vs. saline control), but not 5 or 1 mg/kg doses. Preference for the place paired with 10 mg/kg morphine differed significantly among the studied genotypes (Figure 2A); Kruskal-Wallis one-way ANOVA by genotype, n = 37, χ2 = 7.5, p < .05). Preference for environments paired with 10 mg/kg morphine was eliminated in homozygous knockout transgenic mice, whether on mixed C57/129 (Figure 2A) or MUBX C57BL/6 backcrossed genetic backgrounds (Figure 2B ; Kruskal-Wallis one-way ANOVA by genotype: C57/129 n = 46, χ2 = 10.8, p < .01; MUBX C57BL/6 n = 46, χ2 = 9.4, p < .01).

Place preference conditioning to morphine in μ-opioid receptor knockout mice: C57/129 background. Data represent the mean percent time differences (±SEM) spent in the compartment paired with morphine or saline, comparing pre- and post-conditioning test session for wild-type (+/+), heterozygous (+/−), and homozygous (−/−) μ-opioid receptor genotypes on C57/129 backgrounds. (*p < .05, **p < .01 vs. saline injected group). Mean time spent in the initially non-preferred compartment (sec, ±SEM) during pre-conditioning (Pre) and post-conditioning (Post) sessions were [Wild-type [Pre/Post], heterozygote [Pre/Post], homozygote [Pre/Post]: Saline ([443.3 ± 83.5/449.0 ± 92.0], [320.4 ± 26.3/371.9 ± 124.5}, [350.0 ± 54.1/350.6 ± 65.5]); Morphine 1mg/kg ([320.6 ± 65.2/366.2 ± 72.1], [373.8 ± 90.3/423.5 ± 46.7] (homozygote data missing); Morphine 5mg/kg ([339.7 ± 29.1/413.0 ± 44.6], [353.5 ± 26.0/602.3 ± 71.1] (homozygote data missing); Morphine 10mg/kg ([312.7 ± 41.1/519.6 ± 55.3], [296.0 ± 43.2/649.2 ± 53.2], [315.8 ± 48.5/360.4 ± 41.9]).

Place preference conditioning to morphine in μ-opioid receptor knockout mice: MUBX C57BL/6 background. Data represent the mean percent time differences (±SEM) spent in the compartment paired with morphine (10 mg/kg), comparing pre- and post-conditioning test sessions for wild-type (+/+), heterozygous (+/−), and homozygous (−/−) μ-opioid receptor genotypes on MUBX C57BL/6 backgrounds. (*p < .05 vs. wild-type group). Mean time spent in the initially non-preferred compartment (sec, ± SEM) during pre-conditioning (Pre) and post-conditioning (Post) sessions were [Wild-type [Pre/Post], heterozygote [Pre/Post], homozygote [Pre/Post]: ([444.9 ± 45.1/822.9 ± 52.1], [583.3 ± 127.6/875.4 ± 129.0], [483.8 ± 47.7/597.2 ± 96.7]).
Significant conditioning was observed in heterozygous C57/129 background mice at 5 and 10 mg/kg morphine doses (Figure 2A; p < .05 and .01 vs. saline controls, respectively), whereas 1 mg/kg morphine induced a trend toward place preference (p = .08 vs. saline controls). Heterozygous knockout mice on the C57/129 mixed background displayed almost double the preference for the compartments paired with 5 and 10 mg/kg morphine, when compared to wild-type mice in two replicate experiments (Figure 2A; Kruskal-Wallis One-way ANOVA by morphine dose, n = 49, χ2 = 8.7, p < .05). However, this result was also accompanied by high individual-to-individual variability. In attempts to seek sources of this variability, we tested two additional groups of heterozygous knockout mice on MUBX C57BL/6 backgrounds. One group displayed a strong trend toward enhanced morphine conditioning at the 10 mg/kg dose, but the second group displayed no such trend. Overall, this high variability left the results in MUBX heterozygotes not different from wild-type C57BL/6 mice. No mice that received saline in conditioning sessions exhibited significant alterations in preference for either test compartment. Naloxone pretreatments attenuated preferences induced by 10 mg/kg morphine in both mixed-background wild-type and heterozygous mice (Figure 2C ); p < .01 vs. morphine injected group, eight mice per genotype, Mann-Whitney-Wilcoxon rank sum test).

Place preference conditioning by morphine: Effects of naloxone pretreatment. Data represent the mean percentage differences between the time spent in a compartment in a pre-conditioning test session and the time spent in the compartment after pairing with saline or morphine (10 mg/kg), with or without pretreatment with naloxone (10 mg/kg). Wild-type (+/+) and heterozygous (+/−) μ-opioid receptor genotype mice were tested (*p < .05, **p < .01 vs. saline injected control, # p < .01 vs. morphine injected group). Comparisons of the number of seconds spent in the initially-nonpreferred compartment (±SEM) in pre-conditioning (Pre) and post-conditioning (Post) sessions are displayed as [Pre/Post]: wild-type saline ([443.3 ± 83.5/449.0 ± 92.0], heterozygote saline [320.4 ± 26.3/371.9 ± 124.5]); wild-type saline + naloxone ([443.3 ± 83.5/449.0 ± 92.0], heterozygote saline + naloxone [320.4 ± 26.3/371.9 ± 124.5]); wild-type morphine ([312.7 ± 41.1/519.6 ± 55.3], heterozygote morphine [296.0 ± 43.2/649.2 ± 53.2]); wild-type morphine + naloxone ([312.7 ± 41.1/519.6 ± 55.3], heterozygote morphine + naloxone [296.0 ± 43.2/649.2 ± 53.2]).
Intravenous Self-Administration
Water training Mice of each genotype successfully learned to lever press for water reinforcement so that there were no significant differences between genotypes in the rates of acquiring lever pressing behaviors (data not shown).
Morphine Self-Administration Behavior: Fixed Ratio Four Dose-Effect Curve
Lever pressing for morphine on the FR4 schedule varied as a function of genotype and dose (Figure 3A ). Effects of dose and genotype x dose interactions were significant: Dose (F[3,60] = 6.8, p < .005); Genotype × Dose F[6,60] = 2.4, p < .04. There was significant dose-related change in lever pressing behavior in the wild-type mice (F[3,21] = 5.1, p < .008) and a marginally significant dose related change in heterozygotes (F[3,18] = 2.2, p < .06). Morphine maintained significantly greater behavior than saline at the 0.1 mg/kg/inj dose in the wild-type and heterozygous genotypes. Drug intake (mg/kg) was highest at 1.0 mg/kg/injection (18.1, 13.1, and 12.1 mg/kg in the wild-type, heterozygous and knockout mice, respectively) and lowest at the 0.1 mg/kg/injection dose (3.6, 1.6, and 1.3 mg/kg in the wild-type, heterozygous and homozygous knockout mice, respectively). At all doses, drug-intake was greatest in wild-type mice.

Intravenous morphine self-administration. Fixed ratio 4 lever-pressing behavior as a function of increasing morphine dose per injection in wild-type (+/+), heterozygous (+/−), and homozygous (−/−) μ-opioid receptor genotypes. Each point represents the condition mean (±SEM) over the last three sessions of each condition [n = 8 (+/+), n = 7 (+/−), n = 8 (−/−)]. *p < .05 vs. wild-type group.
Morphine Self-Administration Behavior: PR Performance
Figure 3B contrasts the number of 1.0 mg/kg morphine reinforcements received during the first seven days of FR4 self-administration with the reinforcements received during the next seven days progressive ratio schedule access in wild-type and heterozygous mice. There was a significant effect of schedule (FR vs. PR) on the number of injections obtained under each condition (F[1,12] = 28.2, p < .0002). There was no significant main effect of genotype (p < .12) on the number of injections obtained under each condition. The number of reinforcements obtained during the FR4 portion of this experiment was similar to the number obtained data during dose-effect determinations at 1.0 mg/kg. When access to morphine was switched from FR4 to the PR schedule, the number of reinforcements received by mice of each genotype decreased significantly (wild-type F[1,7] = 19.6, p < .003; heterozygous: F[1,5] = 10.4, p < .02). The average final ratio obtained by mice of each genotype during the progressive ratio schedule was 454 for wild-types and 154 for heterozygotes. Total drug intake declined by comparable amounts when mice of each genotype were switched from FR to PR schedules. Intake declined 31% in wild-type and 34% in heterozygotes.

Number of reinforcements obtained under a fixed-ratio 4 and progressive-ratio schedule of reinforcement. Each bar represents the condition mean (±SEM) of results from eight wild-type and six heterozygote mice. *p < .05 versus fixed-ratio 4 group. No significant differences were found between genotypes.
Antinociceptive Tolerance with Chronic Morphine Treatment
Cumulative dose-response curves for morphine-induced antinociceptive responses in hot-plate and tail-flick tests before and after chronic morphine pretreatments are illustrated in Figure 4 . Subcutaneous injection of morphine produced dose-related antinociception in 55°C hot-plate and 53°C tail-flick tests in wild-type (n = 31) and heterozygous animals on mixed C57/129 backgrounds (n = 32). Tolerance to this antinociceptive effect was studied after chronic twice-daily 3, 5, or 7 day morphine pretreatments using dose regimens ascending from 15 mg/kg by 5 mg/kg increments. Wild-type (Figures 4A and C) and heterozygous (Figures 4B and D) mice showed similar right- and downward shifts in morphine dose-effect curves following 3, 5, or 7 days of ascending-dose morphine pretreatments.

Cumulative dose-response curves for morphine-induced antinociceptive responses in hot-plate (HP) or tail-flick (TF) tests before, during and after chronic morphine treatment. Dose-response relationships for morphine-induced alterations of latencies in 55°C hot-plate tests in mice with wild-type (+/+, A) and heterozygote (+/−, B), or 53°C tail-flick testing in mice with wild-type (+/+, C) and heterozygous (+/−, D) μ-opioid receptor genotypes before and after chronic morphine treatment. Percent maximal possible analgesic effect (“% analgesia”) was calculated as described in Methods. A value of zero means no analgesia. *p < .05 vs. acute morphine treated control values for the dosage in naive mice.
A two-way ANOVA of hot-plate test results revealed significant genotype-dependent effects with time course (wild-type F[3,211] = 73.0, p < .001, heterozygote F[3,211] = 68.6, p < .001), and morphine dose (wild-type F[4,211] = 93.1, p < .001, heterozygote F[4,211] = 48.4, p < .01). Tail-flick tests also revealed significant effects for time course (wild-type F[3,211] = 13.6, p < .001, heterozygote F[3,217] = 34.0, p < .01), and morphine dose (wild-type F[4,211] = 21.2, p < .01, heterozygote F[4,217] = 42.2, p < .01). Scheffe post-hoc tests demonstrated that the fraction maximal antinociception induced by morphine was higher in both naïve wild-type and heterozygous knockout mice than it was in mice pretreated with five or seven days of morphine (p < .05), with data from hot-plate tests using 56 mg/kg morphine in heterozygous mice providing the only exception. The relative morphine potencies in tail-flick and hot-plate assay were similar in wild-type and heterozygous mice (Table 1).
Physical Dependence Revealed by Naloxone-Precipitated Abstinence After Chronic Morphine Treatments
Development of physical dependence was studied after twice-daily morphine treatments for 3, 5, or 7 days with a dose regimen ascending from 15 mg/kg by 5 mg/kg daily increments. When withdrawal was precipitated by naloxone in mice pretreated with morphine for five or seven days, the amount of precipitated jumping did not differ between wild-type (n = 22) and heterozygous knockout animals on mixed C57/129 backgrounds (n = 21) (Figure 5 ). Heterozygous animals tested after 3 days’ morphine treatments revealed trends toward a greater number of precipitated withdrawal signs than those found in wild-type mice (n = 10 and 9, respectively) that did not reach statistical significance (p = .08, Mann-Whitney-Wilcoxon rank sum test). Homozygous knockout mice tested after 5 days’ morphine treatments showed no naloxone-precipitated jumping (n = 7).

Jumping induced by naloxone in mice treated with a chronic morphine regimen. Naloxone-precipitated jumping after chronic morphine treatment in mice with wild-type (+/+), and heterozygous (+/−) μ-opioid receptor genotypes. # p = .08 vs. wild-type.
Morphine Lethality
Lethality was universal among animals of each of the three mixed-background genotypes that received 1000 mg/kg morphine (Figure 6A ). Morphine caused convulsions prior to death in most mice regardless of genotype. There was a gene-dose-dependent shift to the right in LD50 values. These values were approximately 500 mg/kg for wild-type (n = 47), 600 mg/kg for heterozygotes (n = 60), and 700 mg/kg for homozygous knockout mice (n = 24). 750 mg/kg of morphine, a dose lethal to all wild-type littermates, spared 25% of the homozygous knockout mice. Heterozygous animals administered 650 mg/kg morphine showed significantly longer latencies before morphine was lethal than wild-type mice (Figure 6B; ANOVA followed by Scheffe post-hoc analysis: F[2,24] = 10.8, p < .05). The mean latencies prior to lethality at 750 and 1000 mg/kg were not increased significantly in a gene dose-related fashion, however.

(A) Dose-response relationships for morphine mortality. LD50 values for morphine were approximately 500 mg, 600 mg, and 700 mg in wild-type (+/+), heterozygote (+/−) and homozygote (−/−) μ-opioid receptor genotypes, respectively. (B) Latencies for morphine mortality. The loss of μ receptor gene increased mean latencies to death in a gene dose-related fashion. *p < .05 vs. wild-type (+/+).
DISCUSSION
These studies of morphine effects in homozygous μ receptor knockout mice provide additional support for the μ receptor dependence of several of the morphine effects assessed here. Results from heterozygotes indicate differences in apparent μ receptor reserve manifest in circuits producing μ-dependent effects that include reward, locomotion, antinociception, lethality, tolerance, and physical dependence. Interpretation of each observation should also be tempered by considerations of the other neuroadaptive alterations that partial or total μ receptor deletion cause in these mice during their development and adulthood.
Heterozygous mice express virtually half, 56%, of wild-type levels of μ receptor Bmax values when studied by radioligand binding saturation analyses (Sora et al. 1997b). In reverse transcriptase-polymerase chain reaction (RT-PCR) analysis, μ receptor mRNA levels are reduced by at least 50% in heterozygous mice, and are undetectable in homozygotes (I.S. and G.R.U., unpublished observations). These observations contrast with failure to identify any other striking alterations in δ receptors, κ receptors, or expression of preproenkephalin or preprodynorphin mRNAs (I.S. and G.R.U., unpublished observations). The animals do display adaptive alterations in the expression of a number of other genes (Liu et al, 1999). However, only < 0.5% of the genes whose expression can be detected at above-background levels are up- or down-regulated more than 2-fold in these mice on C57/129 mixed genetic backgrounds.
Morphine's interactions with differentially-distributed μ, δ, and κ-opioid receptor populations expressed in different neuronal circuits have been classically invoked to explain different morphine effects. Morphine's lethality in the homozygous knockout mice that lack morphine analgesia (Sora et al. 1997b) could be mediated by the intact δ and/or κ receptor populations found in these animals, or through other mechanisms. The modest alterations in morphine LD50 values induced by removing one μ receptor gene copy and the greater increase in LD50 values in animals with no μ receptor gene copies are both consistent with the idea that morphine's occupancy of μ receptors contributes modestly to moderately to morphine lethality in wild-type mice. No differences in the modes of lethality are apparent. Mice of each genotype died with the cyanosis and poor inspiratory efforts that indicate respiratory depression, accompanied by terminal tonic/clonic epileptiform activities (Brase et al. 1977). Although morphine lethality has been reported to be unchanged in naloxone-treated mice (Pasternak 1981), lethality in homozygous knockout mice could still be caused by opiate- receptor-mediated effects of this alkaloid.
Morphine LD50 values for wild-type mice, 500 mg/kg, are similar to those reported for other strains of mice. LD50 values for heterozygous and homozygous μ receptor knockout mice reported from other laboratories (Loh et al. 1998) were higher than LD50 values from the current work. Subtle differences in testing or differences in genetic backgrounds or testing procedures [129/Ola (Loh et al. 1998) vs. 129SvEv (Sora et al. 1997b)] may explain these differences. Other work does support the idea that μ opioid receptors play only a modest role in morphine lethality. Differences in genetic background and/or testing conditions could also contribute to the modest differences between our locomotor data and that of Tian and coworkers (Tian et al. 1997). These investigators reported reduced baseline spontaneous horizontal activity in μ knockout homozygous mice developed on a mixed genetic background. We did not observe such differences in our mice with the mixed C57/129 genetic background.
Morphine interactions with μ, δ, and κ-opioid receptor populations expressed in different neuronal circuits have been classically invoked to explain varying morphine pharmacologic effects. Morphine-induced reward, locomotion, thermal antinociceptive tolerance and physical dependence are each virtually ablated in homozygous μ receptor knockout mice and are thus strikingly μ receptor dependent. Even such strikingly μ receptor dependent effects, however, are likely to involve distinct brain circuits (Behbehani 1995). These circuits contain distinct neuronal populations that express differing densities of μ receptors (Moriwaki et al. 1996), the Gi and Go class guanine nucleotide binding proteins activated by agonist-occupied μ receptors (Reisine et al. 1996), opiate-modulated second messenger systems (Childers 1993), opiate-modulated potassium (Kovoor et al. 1995) and calcium channels (North 1993), and other effector systems. Differences in the relationships between μ receptor densities and the relative compositions and densities of these other cellular effector proteins could result in circuit-to-circuit differences in the consequences of the heterozygotes’ failure to express normal complements of μ receptors. Such biochemical differences could result in different patterns of μ receptor reserve even for these highly μ dependent morphine effects. Gene-dose effects, manifest in different ways in the heterozygous knockout mice tested here, do appear to suggest that distinct brain circuits that provide different morphine effects may well display different levels of receptor reserve.
If morphine dose-response relationships are shifted right- and downward in heterozygotes, little receptor reserve is evident. These sorts of relationships have been established for heterozygote responses for morphine analgesia, and for morphine-induced locomotor activity stimulation [(Matthes et al. 1996; Sora et al. 1997b), and current data]. Right- and downward shifts in analgesia dose response curves are substantial for heterozygotes and profound for homozygotes. Fixed ratio (FR4) morphine self-administration exhibits a profoundly-downward shifted dose-effect relationship in heterozygotes, consistent with little receptor reserve. Since significant dose-related changes in behavior maintained by morphine injections were still evident in the heterozygous mice, some receptor reserve is evident for both these phenotypes. In fact, if the self-administration data are compared as a percent of the saline response rate, the difference between the heterozygotes and the wild-type mice is approximately 20–30% whereas the difference between these genotypes is much greater in the acute analgesic assay. A quantitative comparison of the receptor reserve difference between phenotypes in this study is difficult given nociceptive stimulus intensity considerations and single point (50% reduction in the heterozygotes) characterizations. However, a previous study using the method of partial irreversible antagonism also suggests a greater receptor reserve for opioid self-administration than opioid-induced analgesia (Zernig et al. 1997).
Circuits in which μ receptors mediate morphine effects with significant receptor reserves might allow full morphine effects with loss of even half of the μ receptor levels found in wild-type mice. Neither morphine-induced tolerance nor the level of physical dependence ultimately attained is significantly affected by the reduced μ receptor densities found in heterozygotes, although there is a slight trend for slower acquisition of full physical dependence evident in the day 3 precipitated abstinence data. Both of these phenomena are absent from homozygous knockout mice, however. Morphine conditioned place preference also suggests significant receptor reserves in mice of mixed C57/129 genetic background, though less consistent reserve in the C57 backcrossed congenic animals. Reductions in the numbers of morphine reinforcements received by heterozygous mice on fixed ratio and progressive ratio schedules are both less than 50% compared to wild-type values. The combined data is consistent with the idea that μ receptor densities and levels of expression of other proteins whose levels could differ between mouse strains could contribute to varying amounts of receptor reserve for different rewarding processes.
Naloxone-precipitated jumping occurs at identical frequencies in heterozygote and wild-type mice tested after five- or seven-day morphine treatments. These observations support a substantial receptor reserve for this robust murine measure of morphine physical dependence. The modest differences in jumping that can be precipitated by naloxone on the third day of morphine treatments in wild-type and heterozygous mice do not reach statistical significance. They do leave open the possibility that the receptor reserve for tolerance might not be much larger than 50%. Morphine-induced tolerance is manifest by down- and rightward shifts in morphine antinociceptive dose-response relationships in both wild-type and heterozygous mice pretreated with multiday morphine regimens. Since heterozygotes display less morphine analgesia without pretreatments than do wild-type mice, the magnitude of the right- and downward shifts in dose-effect relationships assessed in heterozygotes after several days’ morphine pretreatment are smaller than those in wild-type mice. However, the fractional reductions in morphine's antinociceptive efficacies are similar in tests performed after five days’ morphine pretreatment in mice of both genotypes, as the relative potencies in both tail-flick and hot-plate assay did not differ between wild-type and heterozygous mice. These data thus appear to provide evidence for substantial receptor reserves in the brain mechanisms that contribute to morphine tolerance in nociceptive testing.
μ receptor reserves, and cell-to-cell differences in receptor reserves, have been identified in in vitro studies in which pharmacological treatments inactivating varying μ receptor populations can lead to dramatic reductions in some morphine effects while sparing others (Chavkin and Goldstein 1984). The data from these knockout mice can thus be compared to data from mice pretreated in vivo with alkylating agents that can inactivate μ receptors with varying degrees of selectivity. Pretreatments with 2.5 μg of β-funaltrexamine (β-FNA) shifted morphine and levorphanol dose-effect curves to the right, while higher doses of 5.0 μg also reduced the maximum agonist effects (Adams et al. 1990). Tolerance to the analgesic effects of μ opioid receptor agonists can vary with the intrinsic activities both of the drug used to induce the tolerance and the drug whose analgesic actions are assessed (Paronis and Holtzman 1992). However, with the exception of these studies, μ receptor reserve has had little prior direct experimental support in intact animals. Studies of knockout mice with varying levels of μ receptor expression now allow tentative in vivo identification of differences between μ receptor reserve for morphine's actions at one μ receptor-expressing circuit compared to those at another circuit.
No identified μ receptor deficient humans allow assessments of opiate drug effects in totally receptor-deleted individuals. However, significant human inter-individual differences in the levels of expression of μ receptors are well documented in postmortem (Paronis and Holtzman 1992; Gabilondo et al. 1995) and in vivo positron emission tomographic studies of μ receptor densities (Frost et al. 1990). Heterozygous mice with half of wild-type μ receptor densities provide a potential model for the ways in which different levels of μ receptor expression could influence opiate drug responses in humans. Our present results may have implications for morphine effects in human individuals who might express, for example, subnormal complements of μ receptors. If humans behave in fashions similar to those of these mice, individual differences in μ receptor expression levels could lead to significant inter-individual differences in effective analgesic opiate doses in the absence of substantial receptor reserve in such circuits, with less difference in the abilities of opiates to produce tolerance and physical dependence. Each of these considerations suggests that pain might be more difficult to treat with μ agonists in humans with fewer μ receptors.
Although concepts of receptor reserve have largely been developed using in vitro assays in which response strength is assessed in preparations in which previous-existing receptors have been inactivated, the current data suggests substantial circuit to circuit differences in this process in vivo. These considerations could be of importance for design of therapeutic regimens that might minimize addictive and maximize analgesic properties of opiate therapeutics.
References
Adams JU, Paronis CA, Holtzman SG . (1990): Assessment of relative intrinsic activity of mu-opioid analgesics in vivo by using beta-funaltrexamine. J Pharmacol Exp Ther 255: 1027–1032
Aghajanian GK . (1978): Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature 276: 186–188
Beardsley PM, Meisch RA . (1981): A precision drinking device for rats tested with water, etonitazene, and ethanol. Pharmacol Biochem Behav 14: 871–876
Behbehani MM . (1995): Functional characteristics of the midbrain periaqueductal gray. Prog Neurobiol 46: 575–605
Brase DA, Loh HH, Way EL . (1977): Comparison of the effects of morphine on locomotor activity, analgesia and primary and protracted physical dependence in six mouse strains. J Pharmacol Exp Ther 201: 368–374
Chavkin C, Goldstein A . (1984): Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci U S A 81: 7253–7257
Childers SR . (1993): Opioid receptor-coupled second messenger systems. In Herz A (ed), Opioids I, Handbook of Experimental Pharmacology. Berlin, Springer-Verlag, pp 189–216
Dewey WL, Snyder JW, Harris LS, Howes JF . (1969): The effect of narcotics and narcotic antagonists on the tail-flick response in spinal mice. J Pharm Pharmacol 21: 548–550
Di Chiara G, North RA . (1992): Neurobiology of opiate abuse. Trends Pharmacol Sci 13: 185–193
Elmer GI, Pieper JO, Goldberg SR, George FR . (1995a): Opioid operant self-administration, analgesia, stimulation and respiratory depression in mu-deficient mice. Psychopharmacology (Berl) 117: 23–31
Elmer GI, Evans JL, Ladenheim B, Epstein CJ, Cadet JL . (1995b): Transgenic superoxide dismutase mice differ in opioid-induced analgesia. Eur J Pharmacol 283: 227–232
Frost JJ, Mayberg HS, Sadzot B, Dannals RF, Lever JR, Ravert HT, Wilson AA, Wagner HJ, Links JM . (1990): Comparison of [11C]diprenorphine and [11C]carfentanil binding to opiate receptors in humans by positron emission tomography. J Cereb Blood Flow Metab 10: 484–492
Fuchs PN, Roza C, Sora I, Uhl G, Raja SN . (1999): Characterization of mechanical withdrawal responses and effects of mu-, delta- and kappa-opioid agonists in normal and mu-opioid receptor knockout mice. Brain Res 821: 480–486
Funada M, Suzuki T, Sugano Y, Tsubai M, Misawa M, Ueda H, Misu Y . (1994): Role of beta-adrenoceptors in the expression of morphine withdrawal signs. Life Sci 54: PL113–PL118
Furchgott RF . (1967): The pharmacological differentiation of adrenergic receptors. Ann N Y Acad Sci 139: 553–570
Gabilondo AM, Meana JJ, Garcia SJ . (1995): Increased density of mu-opioid receptors in the postmortem brain of suicide victims. Brain Res 682: 245–250
Hosohata Y, Vanderah TW, Burkey TH, Ossipov MH, Kovelowski CJ, Bian D, Sora I, Uhl GR, Zhang X, Rice KC, Roeske WR, Hruby VJ, Yamamura HI, Porreca F . (2000): Delta opioid receptor agonists produce antinociception and [35S]GTPγS binding in mu-opioid receptor knock-out mice. Eur J Pharmacol 388: 241–248
Johnson SM, Fleming WW . (1989): Mechanisms of cellular adaptive sensitivity changes: Applications to opioid tolerance and dependence. Pharmacol Rev 41: 435–488
Kenakin TP . (1984): The classification of drugs and drug receptors in isolated tissues. Pharmacol Rev 36: 165–222
Kieffer BL, Befort K, Gaveriaux RC, Hirth CG . (1992): The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc Natl Acad Sci U S A 89: 12048–12052
Kitanaka N, Sora I, Kinsey S, Zeng Z, Uhl GR . (1998): No heroin or morphine 6beta-glucuronide analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol 355: R1–R3
Kovoor A, Henry DJ, Chavkin C . (1995): Agonist-induced desensitization of the mu opioid receptor-coupled potassium channel (GIRK1). J Biol Chem 270: 589–595
Liu Q-R, Sora I, Walther D, Uhl GR . (1999): Effects of μ opiate receptor deletion on other genes’ expression patterns. Soc Neurosi Abstr 25: 36
Loh HH, Liu HC, Cavalli A, Yang W, Chen YF, Wei LN . (1998): mu Opioid receptor knockout in mice: Effects on ligand-induced analgesia and morphine lethality. Brain Res Mol Brain Res 54: 321–326
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le MM, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL . (1996): Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383: 819–823
Minami M, Toya T, Katao Y, Maekawa K, Nakamura S, Onogi T, Kaneko S, Satoh M . (1993): Cloning and expression of a cDNA for the rat kappa-opioid receptor. Febs Lett 329: 291–295
Miner LL, Drago J, Chamberlain PM, Donovan D, Uhl GR . (1995): Retained cocaine conditioned place preference in D1 receptor deficient mice. Neuroreport 6: 2314–2316
Miner LL, Marley RJ . (1995): Chromosomal mapping of the psychomotor stimulant effects of cocaine in BXD recombinant inbred mice. Psychopharmacology (Berl) 122: 209–214
Mizoguchi H, Narita M, Oji D, Suganuma C, Nagase H, Sora I, Uhl G R, Tseng L F . (1999): The Mu-opioid receptor gene-dose dependent reductions in responses to G protein activation in the pons/medulla and supraspinal antinociceptive actions by endomorphins in mu-knockout mice. Neuroscience 94: 203–207
Moriwaki A, Wang JB, Svingos A, van Bockstaele E, Cheng P, Pickel V, Uhl GR . (1996): mu Opiate receptor immunoreactivity in rat central nervous system. Neurochem Res 21: 1315–1331
Narita M, Mizoguchi H, Sora I, Uhl GR, Tseng LF . (1999): Absence of G-protein activation by mu-opioid receptor agonists in the spinal cord of mu-opioid receptor knockout mice. Br J Pharmacol 126: 451–456
North RA . (1993): Opioid actions on membrane ion channels. In Herz A (ed), Opioids I, Handbook of Experimental Pharmacology. Berlin, Springer-Verlag, pp 773–797
Paronis CA, Holtzman SG . (1992): Development of tolerance to the analgesic activity of mu agonists after continuous infusion of morphine, meperidine or fentanyl in rats. J Pharmacol Exp Ther 262: 1–9
Pasternak GW . (1981): Opiate, enkephalin, and endorphin analgesia: Relations to a single subpopulation of opiate receptors. Neurology 31: 1311–1315
Pasternak GW . (1993): Pharmacological mechanisms of opioid analgesics. Clin Neuropharmacol 16: 1–18
Porreca F, Burks TF . (1993): Supraspinal opioid receptors in antinociception. In Herz A (ed), Opioids II, Handbook of Experimental Pharmacology. Berlin, Springer-Verlag, pp 21–52
Qiu C, Sora I, Ren K, Uhl G, Dubner R . (2000): Enhanced delta-opioid receptor-mediated antinociception in mu-opioid receptor-deficient mice. Eur J Pharmacol 387: 163–169
Reisine T, Law SF, Blake A, Tallent M . (1996): Molecular mechanisms of opiate receptor coupling to G proteins and effector systems. Ann N Y Acad Sci 780: 168–175
Roberts DC, Bennett SA . (1993): Heroin self-administration in rats under a progressive ratio schedule of reinforcement. Psychopharmacology 111: 215–218
Ruffolo RR Jr . (1982): Review important concepts of receptor theory. J Auton Pharmacol 2: 277–295
Sora I, Funada M, Uhl GR . (1997a): The mu-opioid receptor is necessary for [D-Pen2, D-Pen5] enkephalin-induced analgesia. Eur J Pharmacol 324: R1–R2
Sora I, Li XF, Funada M, Kinsey S, Uhl GR . (1999): Visceral chemical nociception in mice lacking mu-opioid receptors: Effects of morphine, SNC80 and U-50,488. Eur J Pharmacol 366: R3–R5
Sora I, Takahashi N, Funada M, Ujike H, Revay R, Donovan D, Miner L, Uhl GR . (1997b): Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A 94: 1544–1549
Stevens KE, Mickley GA, McDermott LJ . (1986): Brain areas involved in production of morphine-induced locomotor hyperactivity of the C57B1/6J mouse. Pharmacol Biochem Behav 24: 1739–1747
Suzuki T, Funada M, Narita M, Misawa M, Nagase H . (1993): Morphine-induced place preference in the CXBK mouse: Characteristics of mu opioid receptor subtypes. Brain Res 602: 45–52
Tian M, Broxmeyer HE, Fan Y, Lai Z, Zhang S, Aronica S, Cooper S, Bigsby RM, Steinmetz R, Engle SJ, Mestek A, Pollock JD, Lehman MN, Jansen HT, Ying M, Stambrook PJ, Tischfield JA, Yu L . (1997): Altered hematopoiesis, behavior, and sexual function in mu opioid receptor-deficient mice. J Exp Med 185: 1517–1522
Uhl GR, Childers S, Pasternak G . (1994): An opiate-receptor gene family reunion. Trends Neurosci 17: 89–93
Wang JB, Imai Y, Eppler CM, Gregor P, Spivak CE, Uhl GR . (1993): Mu opiate receptor: cDNA cloning and expression. Proc Natl Acad Sci U S A 90: 10230–10234
Wise RA . (1996): Neurobiology of addiction. Curr Opin Neurobiol 6: 243–251
Zernig G, Issaevitch T, Broadbear JH, Burke TF, Lewis JW, Brine GA, Woods JH . (1995): Receptor reserve and affinity of mu opioid agonists in mouse antinociception: Correlation with receptor binding. Life Sci 57: 2113–2125
Zernig G, Lewis JW, Woods JH . (1997): Clocinnamox inhibits the intravenous self-administration of opioid agonists in rhesus monkeys: Comparison with effects on opioid agonist-mediated antinociception. Psychopharmacology (Berl) 129: 233–242
Acknowledgements
This work was supported financially by Intramural Research Program, NIDA and DA11185 (GIE). We thank Nobuyuki Takahashi for excellent consultation; Hsin-Fei Liu, Nancy Goodman, and Steven Kinsey for technical assistance; and Linda Kazlo for assistance with the manuscript. We gratefully acknowledge animal care support from the Charles River/Triad animal care support section. Experiments were conducted under protocols approved by the NIDA Animal Care and Use Committee.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Sora, I., Elmer, G., Funada, M. et al. μ Opiate Receptor Gene Dose Effects on Different Morphine Actions: Evidence for Differential in vivo μ Receptor Reserve. Neuropsychopharmacol 25, 41–54 (2001). https://doi.org/10.1016/S0893-133X(00)00252-9
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(00)00252-9
Keywords
This article is cited by
-
Chemistry and synthesis of major opium alkaloids: a comprehensive review
Journal of the Iranian Chemical Society (2021)
-
Non-nociceptive roles of opioids in the CNS: opioids’ effects on neurogenesis, learning, memory and affect
Nature Reviews Neuroscience (2019)
-
Complex formation between the vasopressin 1b receptor, β-arrestin-2, and the μ-opioid receptor underlies morphine tolerance
Nature Neuroscience (2018)
-
The Cholinergic System as a Treatment Target for Opioid Use Disorder
CNS Drugs (2018)
-
Genetic dissociation of morphine analgesia from hyperalgesia in mice
Psychopharmacology (2017)
