
Abstract
Phagocytosis of dying cells is a major homeostatic process that represents the final stage of cell death in a tissue context. Under basal conditions, in a diseased tissue (such as cancer) or after treatment with cytotoxic therapies (such as anticancer therapies), phagocytosis has a major role in avoiding toxic accumulation of cellular corpses. Recognition and phagocytosis of dying cancer cells dictate the eventual immunological consequences (i.e., tolerogenic, inflammatory or immunogenic) depending on a series of factors, including the type of ‘eat me’ signals. Homeostatic clearance of dying cancer cells (i.e., tolerogenic phagocytosis) tends to facilitate pro-tumorigenic processes and actively suppress antitumour immunity. Conversely, cancer cells killed by immunogenic anticancer therapies may stimulate non-homeostatic clearance by antigen-presenting cells and drive cancer antigen-directed immunity. On the other hand, (a general) inflammatory clearance of dying cancer cells could have pro-tumorigenic or antitumorigenic consequences depending on the context. Interestingly, the immunosuppressive consequences that accompany tolerogenic phagocytosis can be reversed through immune-checkpoint therapies. In the present review, we discuss the pivotal role of phagocytosis in regulating responses to anticancer therapy. We give particular attention to the role of phagocytosis following treatment with immunogenic or immune-checkpoint therapies, the clinical prognostic and predictive significance of phagocytic signals for cancer patients and the therapeutic strategies that can be employed for direct targeting of phagocytic determinants.
Similar content being viewed by others

Facts
-
Recognition and clearance of dying cells is affected by the molecular nature, spatiotemporal frame and overall balance of ‘eat me’ and ‘don't eat me’ signals exposed on the surface of dying cells.
-
During carcinogenesis, both cell death and phagocytic clearance mechanisms tend to become inefficient and cooperate to expand premalignant clones that resist antitumour immunity.
-
The mechanisms of cancer cell death elicited by anticancer therapy and the type of phagocytes (e.g., tumour-resident versus therapy-recruited) interacting with dying cells are decisive factors in making a difference between anti-inflammatory or pro-inflammatory responses.
-
At the two extremes of a spectrum, tolerogenic phagocytosis represents a tolerogenic ‘eat me’ signal-dependent engulfment of dying cancer cells that leads to active immunosuppression. On the other hand, immunogenic phagocytosis is an immunogenic ‘eat me’ signal-dependent engulfment of dying cancer cells that facilitates immuno-stimulatory clearance of cancer cell corpses.
Open Questions
-
It is unknown to what extent the mechanisms and/or consequences of phagocytic removal tend to be cell death pathway specific.
-
It is unknown if specific ‘eat me’ signals govern the intracellular processing route of the engulfed cargo and thereby regulate the presentation of cancer antigens.
-
The mechanisms and immunological consequences of immune cell-mediated endocytosis of cellular fragments, microparticles and/or exosomes released from dying cells need urgent characterization in near future.
-
It remains enigmatic whether immune cells showing preimmunosuppressed state can mature or turn immunostimulatory upon immunogenic phagocytosis.
-
For a large majority of FDA-approved anticancer therapies, there is no clarity on specific ‘eat me’ signals or immunological consequences of phagocytosis – this needs further characterization.
-
In the future, it would be crucial to characterize whether immune-checkpoint therapies stimulate antibody-dependent cellular phagocytosis with immunogenic consequences.
-
An important challenge is to develop methodologies to detect active phagocytosis in clinical tumour samples and ascertain its prognostic or predictive impact.
Clearance mechanisms of dying cells
Homeostatic tissue turnover is facilitated by regulated cell death, mainly in the form of apoptosis (a physiological form of cell death; Box 1) that avoids leaking contents and stimulates rapid, immunologically ‘silent’ phagocytic clearance.1, 2, 3 Failure to clear apoptotic corpses causes release of their intracellular components possibly evoking undesired inflammatory responses (e.g., autoimmunity).3, 4 Clearance of dying cells is carried out by both professional phagocytes of the innate immune system (i.e., macrophages (MФ), immature dendritic cells (DCs), neutrophils) and non-professional phagocytes (e.g., epithelial cells in the skin or intestine). However, the professional phagocytes are better adapted at antigen cross-presentation (especially DCs, which are the principle antigen-presenting cells (APCs)).5, 6, 7, 8, 9
To ensure their efficient removal, physiologically dying cells emit ‘find-me signals’ (e.g., fractalkine (CX3CL1)) to recruit anti-inflammatory phagocytes,10 or release ‘keep-out’ signals (e.g., lactoferrin), to avoid inflammatory cells.7, 11, 12 Along with these soluble signals, clearance of dying cells is regulated by a constellation of ‘eat me’ signals, a collective term for surface-tethered proteins, phospholipids or protein complexes facilitating cellular engulfment by binding to phagocytic receptors on immune cells (Figure 1).5, 7, 11, 12 Viable cells avoid phagocytic clearance through retained presentation of surface-associated ‘don't eat me’ signals.6, 13 On the other hand, recognition and clearance of dying cells is affected by the molecular nature, spatiotemporal frame and overall balance of pro-phagocytic and antiphagocytic determinants,14 for example, dying cells tend to reduce ‘don't eat me’ signals while increasing the ‘eat me’ signals on the surface.5, 7, 11, 12 Excessive cell death events can overwhelm the clearance capacity of phagocytes thereby causing a persistence of late apoptotic or secondary necrotic cells capable of disturbing tissue homeostasis.14 Such corpses are cleared through mechanisms that have been partially deciphered15 and involve a complex repertoire of receptors, opsonins and cell-associated ligands.14 Finally, the relevance and contribution of non-apoptotic cell death mechanisms, including various forms of regulated necrosis such as necroptosis (Box 1),16 in tissue homeostasis remains unclear.

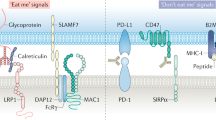
A schematic representation of major ‘eat me’ and ‘don't eat me’ signals regulating phagocytosis of dying cancer cells and the spectrum of subsequent immunological responses. (a) The immunological consequences of phagocytosis of dying cancer cells can be viewed as a spectrum of responses ranging from immunogenic and inflammatory to semi-tolerogenic and tolerogenic. Here immunogenic phagocytosis (induced by ICD inducers or the presence of ADCP plus TLR/TLR agonists) and tolerogenic phagocytosis (induced by non-immunogenic therapies or in basal conditions) occupy the two diametrically opposite poles of this spectrum, and consist of the most resolved immunological responses. On the other hand, inflammatory or semi-tolerogenic phagocytosis may result in context-specific immunological responses that are less resolved and thus more complex to decipher or exploit. (b) Cancer cells dying under basal conditions or following treatment with non-immunogenic therapies undergo tolerogenic phagocytosis mediated by interaction between tolerogenic ‘eat me’ signals (on dying cancer cells) and their respective cognate receptors (on phagocytes). This facilitates immunosuppression driven by anti-inflammatory cytokines. On the other hand, cancer cells dying following treatment with immunomodulatory therapies or inducers of ICD undergo immunogenic phagocytosis mediated by interaction between immunogenic ‘eat me’ signals and their respective cognate receptors. This facilitates immunostimulation driven by pro-inflammatory cytokines. Cancer cell death is also usually accompanied by downregulation of ‘don't eat me’ signals, such as CD47. CD, cluster of differentiation; Gas6, growth arrest-specific 6; ICAM3, intercellular adhesion molecule 3; LRP1, low-density lipoprotein receptor-related protein 1; TGF, transforming growth factor
Besides physiological events, phagocytosis also has a vital role in the control of injured, infected or diseased cells, such that inefficient phagocytosis may exacerbate disease.15 For example, during carcinogenesis the inefficient phagocytosis of dying cancer cells, resulting from the overwhelming of the phagocytic system, may cause the persistence of necrotic cells in tumours (a prominent negative prognostic factor).3, 5, 17, 18 In a therapy-context, cancer cell death can occur through different mechanisms (as discussed later), which in turn can decisively affect the mechanisms of phagocytosis and its immunological consequences.5 However, both cell death and phagocytic clearance mechanisms become inefficient in a tumour-context, thereby cooperating to expand premalignant clones resisting antitumour immunity. In this scenario, the main purpose of anticancer treatments should not only be limited to inducing cancer cell death but should also involve facilitating efficient phagocytosis-based transfer of crucial tumour-associated antigens (TAAs; that include both classical and neo-antigens).3, 5, 17, 18 This would allow processing and presentation of TAAs on the level of effector innate immune cells such as DCs with proper co-stimulation.3, 19, 20 Such activated immune cells can further activate the effector adaptive immune cells (e.g., CD4+ T cells polarized for type-I antitumour immune reactions, that is, interferon (IFN)-γ-producing CD4+ T cells or Th1 cells, and cytotoxic CD8+ T lymphocytes (CTLs)).1, 20 Properly activated T cells are capable of targeting and eliminating the (residual) malignant cells based on TAAs presented to them.3, 19, 20
Thus the nature, intensity and context of phagocytosis in a tumour are pivotally positioned at the interface between cancer cell death and the immune system.3 In the present review, we discuss this pivotal role of phagocytosis in regulating responses to anticancer therapy, in particular, immunogenic and immune-checkpoint therapies. We also discuss the prognostic and predictive significance of ‘eat me’/’don't eat me’ signals for cancer patients and the clinical translation of therapies targeting these signals.
Impact of therapy-induced cancer cell death on phagocytosis
The mechanisms of cancer cell death elicited by anticancer therapy and the type of phagocytes (e.g., tumour-resident versus therapy-recruited) involved in their clearance, are decisive factors between inducing anti-inflammatory responses or TAA-directed immunity.21
In the past decades, compelling evidence has challenged the original simplistic dichotomy that classified apoptosis as a tolerogenic cell death (TCD) and necrosis as a pathological cell death inherently pro-inflammatory/immunogenic (Box 1). Indeed, certain forms of cancer cell apoptosis (termed immunogenic cell death (ICD), Box 1)19 can be perceived as ‘non physiological’ by the immune system, which reacts by engaging an efficient host immune defense.1 ICD triggered by certain anticancer modalities inducing the combined occurrence of reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress19 is highly immunogenic owing to emission of danger signals or damage-associated molecular patterns (DAMPs) and other immunostimulatory molecules (Box 1 lists the known DAMPs/immunomodulatory molecules associated with ICD)21 and is able to elicit T-cell mediated antitumour immunity.1 Based on the main immunological profiles of cancer cell death (i.e., TCD and ICD), the subsequent phagocytic contexts can also be mainly associated with tolerogenic and immunogenic responses (Figure 1). Here tolerogenic phagocytosis can be defined as homeostatic engulfment of dying cancer cells that leads to induction of tolerogenicity (also owing to anti-inflammatory factors released by dying cells, Box 1) (Figure 1). Conversely, immunogenic phagocytosis can be defined as a non-homeostatic engulfment of dying cancer cells19 that causes increased production of pro-inflammatory cytokines/chemokines (also owing to further co-stimulation provided by danger signals21 and/or Toll-like receptor (TLR) agonists released by dying cells, Box 1), resulting in immunostimulatory clearance of cancer cell corpses (Figure 1).1 It is also possible (albeit still poorly characterized) that the immunological consequences of phagocytosis are differentially modulated by the type of phagocytes that are recruited by TCD (anti-inflammatory MФ or neutrophils) or ICD (inflammatory monocytes or specific DCs, for example, CD11c+CD11b+Ly6ChiDCs22 or CD8α+ DCs)1 and the (inflammatory) microenvironment where clearance takes place.
However, it should be noted that tolerogenic phagocytosis and immunogenic phagocytosis represent two extreme polar-ends of the clearance mechanism. In reality, phagocytosis of dying cancer cells can give rise to a spectrum of inflammatory responses, which may be associated with ambiguous immunological reactions23, 24 that can facilitate pro- or antitumorigenic responses in a context-dependent manner (Figure 1).25, 26 Such responses tend to be quite distinct from pure tolerogenic or immunogenic responses on the levels of cytokines, chemokines, DAMPs and balance or misbalance between ‘eat me’ or ‘don't eat me’ signals23, 24, 25, 26 (as detailed more exhaustively elsewhere27, 28, 29). For the sake of clarity and focussed discussion, in this review we will only elaborate upon the two extreme polar-ends of this continuum (i.e., tolerogenic and immunogenic phagocytosis; Figure 1).
Anticancer therapies evoke various cancer cell death mechanisms, which may even coexist. The biological or therapeutic contexts where apoptosis or necrosis can evoke tolerogenicity or immunogenicity have been described.30 However, similar knowledge is seldom available for other cell death pathways2 such as necroptosis, autophagic cell death, mitotic catastrophe, parthanatos and ferroptosis (Box 1) – a gap in knowledge that requires urgent attention. For instance, necroptosis31 and autophagic cell death32 offer a therapeutic alternative to kill apoptosis-resistant cancer cells,33 but recognition and clearance of necroptotic/autophagic cells by phagocytes is not completely understood. It is presumable that the uptake of necroptotic cells involves similar inflammatory mechanisms as applicable to necrosis owing to a resemblance in their terminal morphologies.14, 16, 34 However, it is also possible that kinase-driven signalling events during necroptosis modify cellular components, generating a different immunobiology, for example, immunosuppression.35 However, recent evidence suggests that heightened autophagy in cancer cells can suppress the emergence of immunogenic ‘eat me’ signals, such as surface-calreticulin (ecto-CRT).36, 37, 38 In another context, autophagic dying cells have been found to undergo (phosphatidylserine (PtdSer)-based)8 phagocytosis associated with inflammatory response.39 Thus in the future it would be crucial to identify the molecular determinants driving the recognition and phagocytic removal of cancer cells dying through these non-apoptotic pathways.
Tolerogenic ‘eat me’ signals: from PtdSer to DD1α
Tolerogenic ‘eat me’ signals are predominantly exposed not only by cells dying through TCD or physiological apoptosis but also sometimes by necrotic cells40 (Box 1, Figure 1).29 Differential phagocytosis of disintegrated cells is still a matter of debate although recently F-actin was documented as an engulfment signal for (primary or secondary) necrotic cells, binding Clec9a on CD8α+ DCs.41
The best known of the tolerogenic ‘eat me’ signals is externalized PtdSer (Figure 1).8, 15, 40 PtdSer is a phospholipid that normally faces the inner lumen of the bilayered plasma membrane in living cells. However, during the early apoptotic phases it becomes externalized on the outer leaflet of the plasma membrane owing to the coordinated activity of caspases and scramblases (and inactivation of flippases).29, 42, 43 PtdSer binds a large number of immune receptors in a phagocyte-type- and context-specific manner (please refer to other reviews for further insight29). The pro-phagocytic task of PtdSer is further assisted by the presence of phagocytosis-augmenting bridging molecules, for example, Milk-fat globule-EGF factor VIII (MFG-E8) and Gas6 (Figure 1). Such bridging molecules can also support pro-tumorigenic immune reactions. For example, MFG-E8 promotes tumour progression/invasion by favouring tolerogenic phagocytosis-mediated recruitment of immunosuppressive T regulatory cells (Tregs), which are major inhibitors of antitumour immunity.44
Beyond PtdSer, some other surface membrane moieties act as tolerogenic ‘eat me’ signals in a context-dependent manner (although the exact compositional balance of these with PtdSer is still debatable). These include externalized cardiolipin,45 oxidized low-density lipoproteins, annexin-A1, thrombospondin, complement C1q and changes in membrane glycosylation status or charges (Figure 1).6, 15, 46 Interestingly, in the context of chemotherapy-induced ICD, secretion of annexin-A1 by dying cancer cells followed by its binding to the formyl peptide receptor 1 on DCs, was found to facilitate recruitment of tumour-infiltrating DCs in close vicinity to the dying cancer cells and the formation of dead corpse/DC conjugates, resulting in immunogenic phagocytosis.47 This finding further reinforces the concept that the inflammatory context and array of spatiotemporally exposed/secreted factors by the dying cancer cells govern the ultimate immunological responses.48
The most recent molecule to join the ‘club’ of tolerogenic ‘eat me’ signals is a p53 target, namely, immunoglobulin superfamily receptor death domain 1α (DD1α) (Figure 1).49 Of all the known ‘eat me’ signals, DD1α exhibits the most unique and complex immunoregulatory mechanism. On one hand, the (homophilic) DD1α–DD1α interactions between apoptotic cells and phagocytes help in the uptake of the apoptotic cells. On the other hand, these interactions also inhibit the proliferation of CD4+/CD8+ T cells.49 Moreover, the p53-induced expression of DD1α facilitates apoptotic (cancer) cells’ phagocytosis in a PtdSer-independent manner.49 This establishes DD1α as a major immune checkpoint.
Immunogenic ‘eat me’ signals: the role of surface-exposed CRT and heat shock protein 90 (HSP90)
Immunogenic phagocytosis is mediated by a limited number of known ‘eat me’ signals, namely ecto-CRT50 and surface-HSP90 or ecto-HSP90 (Figure 1), which facilitate antitumour immunity.20, 51, 52 The co-existence of an array of such surface-exposed signals is predominantly elicited by cells dying through ICD (Box 1).3, 53, 54, 55 However, in some contexts, specific chemotherapeutics (e.g., melphalan)56 or targeted therapies (e.g., BRAFV600E inhibitor, vemurafenib)57 that are not bona fide ICD inducers can evoke a partial and specific subset of these ICD-associated ‘eat me’ signals/DAMPs and thereby mediate phagocytic clearance with partial immunogenic properties. A very complex interplay between ER stress (centred on the ER stress sensor, protein kinase RNA-like ER kinase (PERK))58 and ROS helps in trafficking of ecto-CRT through the conventional secretory pathway.1, 59, 60 This core trafficking mechanism displays some degree of plasticity3 and has been found to be also regulated by some pro-apoptotic proteins (BAX/BAK/caspase-8), cytosolic Ca2+ or the unfolded protein response signalling proteins (ERp57/eIF2α), depending on the ICD inducer utilized.1, 59, 60, 61 For more on danger signalling pathways, please refer to other recent reviews.1, 3, 19, 54, 62 On the surface of cancer cells, ecto-CRT tends to dock on either lipid rafts and/or LRP1,50, 59 whereas ecto-HSP90 binds prevalently to LRP1 (Figure 1).63 Interaction of these ‘eat me’ signals with some phagocytic receptors on immune cells (e.g., LRP1) aids in removal of cancer cells undergoing ICD (Figure 1, Box 1).38, 52, 64 Ecto-CRT elicits the production of pro-inflammatory cytokines, such as interleukin (IL)-6 and tumour necrosis factor-α (TNF-α) from DCs, thereby facilitating Th1 and/or Th17 polarization.65,36 Moreover, overall expression of CRT mRNA (CALR) in tumour tissue samples (derived from ovarian or lung cancer patients treated with paclitaxel or radiotherapy, respectively) linearly correlates with the levels of genes coding for phagocytosis-related proteins (involved in phagosome maturation or degradation).52 In fact, dying cancer cells naturally incapable of presenting ecto-CRT (owing to an intrinsic resistance mechanism) fail to mediate an anticancer vaccination effect.52 Similarly, HSP90–CD91 binding on immune cells facilitates DC maturation and Th1/17 priming.65 In some contexts, ecto-HSP90 and ecto-CRT are interchangeable in mediating immunogenicity;66 while in other cases, ecto-CRT is the superior immunogenic signal.56 In fact, an in silico analysis suggests that CRT (but not HSP90) possesses close homologues of crucial phagocytosis-assisting motifs.61 Also, ecto-CRT may correlate better with a phagocytosis increase than ecto-HSP90 in the context of anthracycline-induced ICD.67
Surface CD47: a ubiquitous ‘don't eat me’ signal?
A number of ‘don't eat me’ signals have been characterized that act in a context-dependent manner (with the context being type of tissue, type of cells or type of phagocytes).13, 29 However, evidence over time has characterized CD47 as a rather ubiquitous ‘don't eat me’ signal (Figure 1). The binding of CD47 to the immune-receptor signal regulatory proteins α (SIRPα) on phagocytes, inhibits the phagocytosis of CD47-proficient cells.13 Thus, not surprisingly, CD47-deficient cells are critically sensitive to phagocytic clearance.68, 69, 70, 71 Concerning cell death, there are two prevailing models that explain CD47’s antiphagocytic functions. The most widely accepted model entails downregulation of CD47 paralleled by upregulation of ‘eat me’ signals (Figure 1).50 The second model entails spatial repositioning of CD47 away from ‘eat me’ signals.50, 71 CD47 is abundantly overexpressed on cancer cells (especially on cancer stem cells) belonging to various cancer types,72 representing a potent strategy for immune evasion. Moreover, CD47–SIRPα interaction and subsequent SIRPα signalling restricts the efficacy of cancer therapeutic antibodies.73 Beyond SIRPα, CD47 can also interact with some integrins or thrombospondins to modulate IgG antibody-mediated phagocytosis and other inflammatory responses.74, 75, 76 On the level of cancer cells, a HIF1α target protein BNIP3 has been found to regulate CD47 expression levels;77 however, further clarity on CD47-regulating signalling pathway is urgently needed, as it is not entirely known how cancer cell death links with CD47 downregulation or re-localization. Nevertheless, CD47 forms a formidable barrier against cancer cell clearance and thus represents an interesting therapeutic target.
Antibody-dependent cellular phagocytosis: bypassing the ‘eat me’/‘don't eat me’ signals’ interplay?
Phagocytes possess Fcγ receptors (FcγRs) through which they interact with the Fc regions of antibodies to further exert antigen-specific effector functions.78 Interestingly, FcγRs can also mediate antibody-dependent cellular phagocytosis (ADCP) that bypasses the need for canonical phagocytic determinants. More specifically, predominantly type I FcγRs on macrophages or DCs can help in phagocytosis of targets bound to antibodies or antibody complexes (mainly IgG antibodies).78 Such IgG-bound target cells can be efficiently processed and the resulting TAAs can be used for cross-presentation by APCs, thereby enhancing cancer antigen-directed CD4+/CD8+ T-cell responses.78 Importantly, while ADCP proceeds through interactions with type I FcγRs alone, the subsequent immunogenic consequences of such uptake are more tightly governed. In particular (especially in DCs), the activating effects of type I FcγRs are balanced by the inhibitory FcγRIIb receptors,78 which are overcome only if phagocytosis of target cells happens in the presence of additional co-stimulatory signals (e.g., TLR ligands).78 This latter point shows that ADCP might have immunogenic consequences only if the cancer cells die through ICD or necrosis, cell death routines known to release danger signals, including TLR agonists.
Tolerogenic and immunogenic consequences of phagocytic clearance
Besides the nature or balance of the ‘eat me’ signals, the differentiation state of phagocytes can also be a critical factor in defining immunological consequences. In general, tolerogenic ‘eat me’ signals interacting with immature APCs and/or APCs exhibiting immunosuppressive phenotypes (e.g., M2 MФ, N2 neutrophils, myeloid-derived suppressor cells or MDSCs)79 might favour tolerogenic phagocytosis (Figure 1).20, 25 Instead, immunogenic ‘eat me’ signals interacting with immature APCs might favour immunogenic phagocytosis (Figure 1).20, 25 It is unclear whether APCs showing a preimmunosuppressed state (e.g., M2 MФ, MDSCs) can mature upon immunogenic phagocytosis; however, based on available literature this is plausible.80 Of note, ‘eat me’ signals alone are not exclusive immunological determinants as their exposure is invariably accompanied by the emission of other signals (e.g., DAMPs or immunosuppressive cytokines/chemokines).3, 19, 53 Thus, APC’s commitment to tolerogenicity or immunogenicity is regulated by a complex program integrating a variety of signals (Figure 1).
APCs performing phagocytosis (and ADCP) eventually prime the T cells for respective TAAs81 (owing to innate programming of APCs, which constantly process and present any captured antigens to the T cells).82, 83 However, APCs that carry out tolerogenic phagocytosis fail to reach functional maturation and thus present TAAs to CD4+ T cells in the absence of proper co-stimulatory signals (e.g., surface CD80/CD86/CD40/CD83) but possibly in the presence of immunosuppressive cytokines (IL-10/TGF-β), ultimately facilitating the formation of immunosuppressive Tregs (overexpressing immune-inhibitory CTLA-4/PD-1).9, 25, 84, 85, 86 Treg cells not only fail to attack the cancer cells, as their immunosuppressive phenotype categorizes them as ‘safe’/‘self’,9, 86, 87 but also actively secrete pro-tumorigenic cytokines (e.g., IL-6/TNF) and directly eliminate CTLs (through FasL or TRAIL expression).9, 85, 86 Also, APCs that have carried out tolerogenic phagocytosis facilitate cancer progression by disrupting the cross-talk between CD4+ T cells and CTLs. More specifically, tolerogenic APCs present TAAs only to CTLs but not to CD4+ T cells, thereby causing sub-optimal CTL activation.9 Eventually, if re-exposed to TAAs, such CTLs may orchestrate a deranged cytotoxic response that also targets the CD4+ T cells (through TRAIL), thereby facilitating tolerance in the long run.9 Beyond TAA presentation, tolerogenic phagocytosis actively suppresses the secretion of pro-inflammatory cytokines and causes an exaggerated polarization of MФ into a pro-tumorigenic phenotype (owing to the production of pro-tumorigenic cytokines TGF-β and IL-10)88, while promoting the production of anti-inflammatory factors.5, 7, 11, 12, 88
Instead, APCs that carry out immunogenic phagocytosis present TAAs to CD4+ T cells in the presence of heightened levels of co-stimulatory molecules and increased levels of pro-inflammatory cytokines (e.g., IL-6/IL-12/IL-1β) (Figure 1).25, 52, 55 This, in concert, facilitates the differentiation of Th1 cells that orchestrate a type-I immunity-based anticancer programme (consisting of IFN-γ-driven cancer-directed cytostatic effects and suppression of Treg differentiation).19, 36, 89, 90 Simultaneously, these immunogenic APCs allow a productive cross-talk between Th1 cells and CTLs and thereby facilitating CTL-elicited malignant cell elimination (mediated through IFN-γ, FasL–CD95 interaction and perforin-granzyme action) (Figure 2).19, 90

Therapeutic exploitation of phagocytosis of dying cancer cells for T-cell-mediated cancer cell elimination. Treatment with antibody-based anticancer therapies induces FcγR-mediated, ADCP of cancer cells that facilitates antitumour immunity in the presence of co-stimulatory signals such as TLR agonists. Similarly, treatment with ICD-inducing anticancer therapies induces immunogenic phagocytosis driven by immunogenic ‘eat me’ signals or immunogenic DAMPs, components that eventually facilitate antitumour immunity. On the other hand, under basal conditions or after treatment with non-immunogenic anticancer therapies, dying cancer cells undergo tolerogenic phagocytosis that tends to inhibit antitumour immunity by facilitating immunosuppression. Despite these distinct mechanisms and immunological consequences, all three scenarios result in the transfer of cancer antigens (‘classical’ or mutational neo-antigens) from dying cancer cells to the phagocytes. The immunosuppression propagated by tolerogenic phagocytosis can be reversed by treatment with ICT. Antitumour immunity resulting from these treatment scenarios is expected to culminate into cancer cell-eliminating activity exerted by CTLs through IFN-γ (exerts cytostatic effects and polarizes immune cells towards type I-immune reactions), FasL–CD95 interactions (exerts extrinsic apoptosis) or granzyme-perforins secretion (exerts direct cytotoxicity through perforin-driven membrane-pore formation followed by granzyme-induced cell death)
Phagocytic clearance by anticancer therapies
Clinical anticancer therapies that either augment immunogenic potential of cancer cells (e.g., through ICD) or facilitate ADCP in the presence of co-stimulatory ligands are most likely to encourage immunogenic phagocytosis and thus warrant urgent identification (Figure 2). However, clinical therapies that encourage TCD, although less preferable, also need to be identified in order to design smart combinatorial strategies (Figure 2).
To gain a wider view on this important point, we carried out a survey of PubMed publications to ascertain what was known about the immunological consequences of phagocytosis associated with various anticancer therapies (including several FDA-approved ones) (Table 1). To our dismay, only a handful of FDA-approved drugs had some clarity on specific ‘eat me’ signals or immunological consequences of phagocytosis (Table 1). Among these, some agents, such as tamoxifen, sorafenib, bevacizumab, vinblastine and vincristine, exhibited the unfavourable activity of directly inhibiting phagocytic activity of APCs, while others tended to divide between three phagocytic profiles, that is, tolerogenic or immunogenic phagocytosis and ADCP (Table 1).
Harnessing immunogenic phagocytosis via immunogenic anticancer therapies
A number of major anticancer therapies can induce ICD associated with immunogenic phagocytosis-driven anticancer immunity.1 Known ICD inducers, as Table 1 details, include some chemotherapeutics, photodynamic therapy (PDT), radiotherapy, some oncolytic viruses and some physical therapies (Figure 2).1 The immunogenic ‘eat me’ signal mostly characterized for these ICD inducers and confirmed through blockade or intervention strategies (at least for anthracyclines, oxaliplatin, bortezomib, hypericin-PDT, radiotherapy; Table 1) is ecto-CRT. Of note, while many of these anticancer therapies induce apoptotic ICD, it has also emerged that ICD can be necroptotic if induced by the oncolytic Newcastle disease virus.91 It will be important to discover more ICD inducers capable of eliciting a high diversity of immunogenic ‘eat me’ signals.
Encouraging immunogenic phagocytosis via antibody-based immunotherapies
Various anticancer therapeutic antibodies induce ADCP (Table 1), such as rituximab (anti-CD20 antibody), trastuzumab (anti-HER2 antibody) and cetuximab (anti-EGFR antibody). Rituximab and trastuzumab can induce FcγR-mediated anticancer immunity possibly through immunogenic phagocytosis.92 Some clinical observations suggest a correlation between FcγRIIIa (CD16) or FcγRIIa (CD32) polymorphisms and a response to rituximab, trastuzumab and cetuximab.93 Moreover, few of these therapies can liberate co-stimulatory signals from cancer cells (required to make ADCP immunogenic), for example, rituximab causing release of the TLR agonist, HMGB1.94, 95 Overall, these results show that besides their targeted activities, ADCP can help antibody-based therapies to achieve the desirable ‘off-target’ induction of antitumour immunity (Figure 2).93 This raises similar precedence for other immunotherapies targeted towards the cancer cells, that is, anti-PD-L1 antibodies. This is further supported by recent observations of FcγRs modulating the activity of the PD-1/PD-L1 axis.96 In the future, it would be crucial to characterize better the ADCP-associated immunogenic consequences of these antibody-based therapies.
Tolerogenic phagocytosis: foundation for eventual responses to immune-checkpoint therapy (ICT)?
As TAAs are ultimately transferred from cancer cells to the APCs during tolerogenic phagocytosis, does this TAA transfer matter? If so, can the immunosuppressive consequences be reversed? Recent evidence emerging from the ICTs51 suggests that such immunosuppressive consequences can still be reversed (Figure 2). ICTs are therapeutic agents that target regulatory pathways in T cells (e.g., CTLA-4 or PD-1) to enhance antitumour immunity.97, 98, 99 ICTs, however, do not induce a de novo immune reaction,97, 98, 99 since they simply block the immunosuppressive molecules on preexisting T cells already primed for TAAs.97, 98, 99 It is presumable that most of this initial TAA priming of T cells occurred through APCs performing tolerogenic phagocytosis in the tumour microenvironment (Figure 2).97, 98, 99 In the future, it would be crucial to find new targets for ICTs that are exploited by tolerogenic phagocytosis.98, 99 Moreover, considering that currently many clinically applied anticancer therapies tend to induce TCD, and thereby facilitate tolerogenic phagocytosis (Table 1),30 it would be crucial to combine these with ICTs (Figure 2).100
Clinical applications of pro-phagocytic and antiphagocytic determinants
Beyond therapeutic induction of immunogenic phagocytosis/ADCP or reversing the consequences of tolerogenic phagocytosis through ICTs, phagocytic clearance of cancer cells can be more directly exploited for clinical benefits (Figure 3). In the following subsections, these direct applications of pro-phagocytic and antiphagocytic determinants are discussed in further details (Figure 3).

Clinical exploitation of phagocytic determinants for therapeutic targeting or biomarker-driven patient treatment/management. (a) Phagocytosis determinants (i.e., ‘eat me’ signal such as calreticulin (CALR) or ‘don't eat me’ signal such as CD47) can be used as prognostic or predictive biomarkers for stratification or segregation of cancer patients into different risk groups allowing further decision-making regarding specific treatment options. For instance, overall tumour-associated expression levels of CD47 and CALR can be used in synchrony to segregate patients in different treatment groups who would eventually receive different treatments or combinations thereof involving ICD inducers, ICTs and/or anti-CD47 therapy. (b) Phagocytic determinants can also be more directly exploited to therapeutic ends, for example, by targeting through antibodies (against CD47, MFG-E8 or PtdSer) and/or by exogenously providing recombinant versions (e.g., recombinant calreticulin or rCRT), as applicable. CD, cluster of differentiation
‘Eat me’ or ‘don't eat me’ signals as prognostic or predictive biomarkers in cancer
Prognostic and predictive biomarkers, especially those that can be detected in human tumour samples, are valuable for patient management and clinical decision-making (Figure 3a).101, 102, 103, 104 It is technically challenging to detect surface localization of phagocytic determinants in tumour tissue samples90 – a hurdle that has hampered research in this direction. This is particularly important as elevated surface presence of immunogenic ‘eat-me’ signals, such as CRT, should be distinguished from their overall intracellular expression, which is often elevated as a result of stress adaptation and leads to increased cancer cell resistance.105 However, in the right context, the overall expression of the respective molecules can be utilized for patient prognostic analysis or to predict therapy response.90 Table 2 summarizes the prognostic or predictive impact of major phagocytic determinants, among which CRT/CALR and CD47 are the most studied (Table 2). High expression of CD47 is a definitive negative prognostic factor across various cancer types (Table 2),106 whereas the overall picture is much more complex for ecto-CRT/CALR.106 Increased ecto-CRT or high CALR levels predict positive responses to immunogenic anticancer therapies, such as anthracyclines, radiotherapy, paclitaxel and DC vaccines (Table 2).106 However, as a prognostic factor the utility of CALR is limited to only a few cancer types.90 This discrepancy could be because of differences in phagocytic context. In a prognostic biomarker set-up, no differentiation is made between treated or untreated patients, thereby meaning that tumour tissues with both tolerogenic and immunogenic phagocytosis might be tested for CALR levels, thereby confounding the ultimate prognostic impact. On the other hand, in a predictive biomarker set-up, a clear distinction is made between treated and untreated patients.106 This would explain why in contexts of immunogenic anticancer therapies (where immunogenic phagocytosis is likely) high CALR/ecto-CRT levels are positive predictive factors.52 Indeed, in this context, CALR levels tend to positively correlate with levels of phagocytosis-related genes.52 We propose that CD47 and CALR could be used in synchrony for efficient stratification of high- or low-risk cancer patients and for further decision-making regarding the choice of anticancer therapy to be given, as depicted in Figure 3a.106
It is clear that very few phagocytic determinants have been tested so far as prognostic or predictive biomarkers and more studies are needed to reach further clarity. Another challenge would be to detect active phagocytosis in human tumour tissue and directly detect its prognostic or predictive impact.
Combinatorial therapy with recombinant immunogenic ‘eat me’ signals in cancer
Chemotherapeutic ICD inducers cannot be integrated in clinical cell-based vaccination protocols owing to their residual amounts being capable of exerting side effects or toxicity.107 This is one of the primary reasons why clinical anticancer vaccines (whole-tumour-cell or DC vaccines) utilize physicochemical cancer cell death inducers.108 However, while some physicochemical strategies can induce ICD (e.g., radiotherapy, PDT) yet certain others cannot (e.g., freeze/thawing-based necrosis).1, 52, 59, 109 In the latter cases, exogenous addition of recombinant immunogenic ‘eat me’ signals (such as recombinant CRT)52, 56, 64, 110, 111 can complement immunogenic phagocytosis (Figure 3b). In line with this, several studies have used recombinant CRT to augment the immunogenicity of otherwise low immunogenic cancer vaccines (Figure 3b),52, 56, 64, 110, 111 or to promote the immunogenic potential of cancer cells treated with loco-regionally applied chemotherapeutics, such as melphalan.56 Melphalan is a genotoxic drug used often for treatment of limb-confined melanoma through an isolated-limb perfusion/infusion (ILP/ILI) procedure, which involves shunting the limb circulation in order to allow high-concentration melphalan treatment for a limited time followed by its withdrawal from circulation.112, 113 This raises a further prospect of administering the cancer patients with melphalan plus recombinant CRT in an ILP/ILI set-up. This is an exciting prospect that could be safer than systemic treatment (which has autoimmunity-related concerns).7
Combinatorial therapy involving blockade of ‘don’t eat me’ or tolerogenic signals in cancer
Direct blockade of a ‘don’t eat me’ signal (CD47) or a tolerogenic ‘eat-me’ signal (PtdSer)114 are interesting strategies to combine with anticancer therapies/ICTs (Figure 3b). Anti-CD47 antibodies can achieve durable tumour regression in preclinical settings.13, 46, 72 Interestingly, anti-CD47 blockers can synergize with rituximab/trastuzumab in increasing cancer cell clearance and preclinical tumour regression.46 Moreover, while initially anti-CD47 therapy was presumed to mainly involve macrophages, recently it was reported to also activate DC-based priming of antitumour T cells.115 All these promising preclinical results have paved the way for multiple clinical trials with anti-CD47 monoclonal antibodies (Figure 3b), whose results are eagerly awaited (NCT02216409, NCT02367196, NCT02447354, NCT02488811).116 PtdSer can also be a negative prognostic factor (Table 2) and thus an attractive therapy target (Figure 3b).43 In fact, an anti-PtdSer therapeutic antibody, that is, bavituximab has yielded positive results (improved progression-free and overall survival) in a Phase II trial involving lung carcinoma/NSCLC patients.98 Based on these encouraging results and FDA approval, a Phase III trial of bavituximab as a second-line therapy is currently underway for lung carcinoma.98 Preclinical studies have recently also shown that anti-PtdSer can synergize with ICTs to exert antitumour effects.98 Last but not least, pro-phagocytic-bridging molecules can also be therapeutically targeted (Figure 3b). For instance, systemic MFG-E8 blockade increases the effectiveness of conventional chemo-radiotherapy and anticancer vaccines by augmenting apoptosis and potentiating DC-driven immunity.117
Conclusions
The process of phagocytosis was discovered more than a century ago, much before the finer details of cell death regulation and mechanisms came to be described. Despite this, phagocytosis and cell death research have not progressed in synchrony and it is only in the past decade that finer details of cell death pathway-specific phagocytic mechanisms have emerged. Most researchers recognize that the currently known ‘eat me’ and ‘don’t eat me’ signals are only a fraction of what might exist; however, discovery of new phagocytic determinants has been slow. This has also further affected research on immunological consequences of phagocytosis. Although the differentiation between tolerogenic and immunogenic phagocytosis is now starting to emerge, much remains to be resolved. Very few immunogenic ‘eat me’ signals have been discovered. During ICD, while both immunogenic and tolerogenic ‘eat me’ signals co-exist, it still remains unclear how the immunogenic ones ultimately supersede the effects of the tolerogenic ones. Last but not least, not enough FDA-approved therapies have been associated with relevant ‘eat me’ signals, thereby hampering knowledge on the immunogenic or tolerogenic consequences of such therapies. With the clinical success of cancer immunotherapy, it is imperative that more research is carried out on dying cancer cells’ phagocytosis as this is the major route for ordered acquisition of cancer antigens.
Abbreviations
- ADCP:
-
antibody-dependent cellular phagocytosis
- APC:
-
antigen-presenting cell
- CD:
-
cluster of differentiation
- CRT:
-
calreticulin
- CTL:
-
cytotoxic CD8+ T lymphocyte
- DAMP:
-
damage-associated molecular pattern
- DC:
-
dendritic cell
- DD1α:
-
death domain 1α
- Ecto:
-
surface exposure
- ER:
-
endoplasmic reticulum
- HSP:
-
heat shock protein
- ICD:
-
immunogenic cell death
- ICT:
-
immune-checkpoint therapy
- IFN:
-
interferon
- IL:
-
interleukin
- MФ:
-
macrophages
- PDT:
-
photodynamic therapy
- PtdSer:
-
phosphatidylserine
- ROS:
-
reactive oxygen species
- SIRPα:
-
signal regulatory proteins α
- TAA:
-
tumour-associated antigen
- TCD:
-
tolerogenic cell death
- TLR:
-
Toll-like receptor
- Treg:
-
T regulatory cell
References
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol 2015; 6: 588.
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015 22: 58–73.
Garg AD, Martin S, Golab J, Agostinis P . Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ 2014; 21: 26–38.
Griffith TS, Ferguson TA . Cell death in the maintenance and abrogation of tolerance: the five Ws of dying cells. Immunity 2011; 35: 456–466.
Ravichandran KS . Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity 2011; 35: 445–455.
Gregory CD, Brown SB . Apoptosis: eating sensibly. Nat Cell Biol 2005; 7: 1161–1163.
Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P . Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta 2010; 1805: 53–71.
Petrovski G, Zahuczky G, Katona K, Vereb G, Martinet W, Nemes Z et al. Clearance of dying autophagic cells of different origin by professional and non-professional phagocytes. Cell Death Differ 2007; 14: 1117–1128.
Green DR, Ferguson T, Zitvogel L, Kroemer G . Immunogenic and tolerogenic cell death. Nat Rev Immunol 2009; 9: 353–363.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286.
Yang D, de la Rosa G, Tewary P, Oppenheim JJ . Alarmins link neutrophils and dendritic cells. Trends Immunol 2009; 30: 531–537.
Zitvogel L, Kepp O, Kroemer G . Decoding cell death signals in inflammation and immunity. Cell 2010; 140: 798–804.
Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2010; 2: 63ra94.
Poon IK, Hulett MD, Parish CR . Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 2010; 17: 381–397.
Birge RB, Ucker DS . Innate apoptotic immunity: the calming touch of death. Cell Death Differ 2008; 15: 1096–1102.
Krysko DV, Vandenabeele P . Clearance of dead cells: mechanisms, immune responses and implication in the development of diseases. Apoptosis 2010; 15: 995–997.
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T . Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14: 135–146.
Schumacher TN, Schreiber RD . Neoantigens in cancer immunotherapy. Science 2015; 348: 69–74.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L . Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31: 51–72.
Garg AD, Dudek AM, Agostinis P . Cancer immunogenicity, danger signals, and DAMPs: what, when, and how? Biofactors 2013; 39: 355–367.
Venereau E, Ceriotti C, Bianchi ME . DAMPs from cell death to new life. Front Immunol 2015; 6: 422.
Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013; 38: 729–741.
Aderem A . Phagocytosis and the inflammatory response. J Infect Dis 2003; 187 (Suppl 2): S340–S345.
Maderna P, Godson C . Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim Biophys Acta 2003; 1639: 141–151.
Dudek AM, Martin S, Garg AD, Agostinis P . Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol 2013; 4: 438.
Garg AD, Dudek-Peric AM, Agostinis P . Melphalan, antimelanoma immunity, and inflammation-response. Cancer Res 2015; 75: 5400–5401.
Arandjelovic S, Ravichandran KS . Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 2015; 16: 907–917.
Krysko DV, Vandenabeele P . From regulation of dying cell engulfment to development of anti-cancer therapy. Cell Death Differ 2008; 15: 29–38.
Segawa K, Nagata S . An apoptotic 'eat me' signal: phosphatidylserine exposure. Trends Cell Biol 2015; 25: 639–650.
Derer A, Deloch L, Rubner Y, Fietkau R, Frey B, Gaipl US . Radio-immunotherapy-induced immunogenic cancer cells as basis for induction of systemic anti-tumor immune responses - pre-clinical evidence and ongoing clinical applications. Front Immunol 2015; 6: 505.
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014; 343: 1357–1360.
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB . Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15: 171–182.
Fulda S . Caspase-8 in cancer biology and therapy. Cancer Lett 2009; 281: 128–133.
Krysko DV, D'Herde K, Vandenabeele P . Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006; 11: 1709–1726.
Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC et al. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ 2015; 22: 1313–1327.
Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy 2013; 9: 1292–1307.
Verfaillie T, van Vliet A, Garg AD, Dewaele M, Rubio N, Gupta S et al. Pro-apoptotic signaling induced by photo-oxidative ER stress is amplified by Noxa, not Bim. Biochem Biophys Res Commun 2013; 438: 500–506.
Garg AD, Dudek AM, Agostinis P . Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis 2013; 4: e826.
Petrovski G, Ayna G, Majai G, Hodrea J, Benko S, Madi A et al. Phagocytosis of cells dying through autophagy induces inflammasome activation and IL-1beta release in human macrophages. Autophagy 2011; 7: 321–330.
Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell 2004; 15: 1089–1100.
Geijtenbeek TB . Actin' as a death signal. Immunity 2012; 36: 557–559.
Lauber K, Keppeler H, Munoz LE, Koppe U, Schroder K, Yamaguchi H et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ 2013; 20: 1230–1240.
Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med 2004; 200: 1157–1165.
Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G . MFG-E8-mediated uptake of apoptotic cells by APCs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest 2007; 117: 1902–1913.
Balasubramanian K, Maeda A, Lee JS, Mohammadyani D, Dar HH, Jiang JF et al. Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Sci Signal 2015; 8: ra95.
Poon IK, Lucas CD, Rossi AG, Ravichandran KS . Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 2014; 14: 166–180.
Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015; 350: 972–978.
Yang Y, Li XJ, Chen Z, Zhu XX, Wang J, Zhang LB et al. Wogonin induced calreticulin/annexin A1 exposure dictates the immunogenicity of cancer cells in a PERK/AKT dependent manner. PLoS One 2012; 7: e50811.
Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015; 349: 1261669.
Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005; 123: 321–334.
Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE et al. Classification of current anticancer immunotherapies. Oncotarget 2014; 5: 12472–12508.
Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P . Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015; 6: 26841–26860.
Garg AD, Dudek-Peric AM, Romano E, Agostinis P . Immunogenic cell death. Int J Dev Biol 2015; 59: 131–140.
Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P . Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12: 860–875.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3: e955691.
Dudek-Peric AM, Ferreira GB, Muchowicz A, Wouters J, Prada N, Martin S et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res 2015; 75: 1603–1614.
Martin S, Dudek-Peric AM, Maes H, Garg AD, Gabrysiak M, Demirsoy S et al. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in Vemurafenib-resistant melanoma cells. Biochem Pharmacol 2015; 93: 290–304.
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 2012; 19: 1880–1891.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31: 1062–1079.
Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009; 28: 578–590.
Garg AD, Krysko DV, Vandenabeele P, Agostinis P . Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol Immunother 2012; 61: 215–221.
Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G . Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res 2010; 16: 3100–3104.
Gopal U, Bohonowych JE, Lema-Tome C, Liu A, Garrett-Mayer E, Wang B et al. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS One 2011; 6: e17649.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61.
Pawaria S, Binder RJ . CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat Commun 2011; 2: 521.
Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD et al. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res 2010; 70: 9062–9072.
Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res 2011; 71: 4821–4833.
Oldenborg PA . CD47: a cell surface glycoprotein which regulates multiple functions of hematopoietic cells in health and disease. ISRN Hematol 2013; 2013: 614619.
Olsson M, Bruhns P, Frazier WA, Ravetch JV, Oldenborg PA . Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood 2005; 105: 3577–3582.
Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol 2005; 174: 2004–2011.
Nilsson A, Oldenborg PA . CD47 promotes both phosphatidylserine-independent and phosphatidylserine-dependent phagocytosis of apoptotic murine thymocytes by non-activated macrophages. Biochem Biophys Res Commun 2009; 387: 58–63.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009; 138: 286–299.
Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA et al. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci USA 2011; 108: 18342–18347.
Brown E, Hooper L, Ho T, Gresham H . Integrin-associated protein: a 50-kD plasma membrane antigen physically and functionally associated with integrins. J Cell Biol 1990; 111 (Pt 1): 2785–2794.
Cooper D, Lindberg FP, Gamble JR, Brown EJ, Vadas MA . Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc Natl Acad Sci USA 1995; 92: 3978–3982.
Lindberg FP, Bullard DC, Caver TE, Gresham HD, Beaudet AL, Brown EJ . Decreased resistance to bacterial infection and granulocyte defects in IAP-deficient mice. Science 1996; 274: 795–798.
Maes H, Van Eygen S, Krysko DV, Vandenabeele P, Nys K, Rillaerts K et al. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis 2014; 5: e1127.
Bournazos S, Ravetch JV . Fcgamma receptor pathways during active and passive immunization. Immunol Rev 2015; 268: 88–103.
Kersten K, Salvagno C, de Visser KE . Exploiting the immunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front Immunol 2015; 6: 516.
Lin TJ, Lin HT, Chang WT, Mitapalli SP, Hsiao PW, Yin SY et al. Shikonin-enhanced cell immunogenicity of tumor vaccine is mediated by the differential effects of DAMP components. Mol Cancer 2015; 14: 174.
Brusa D, Garetto S, Chiorino G, Scatolini M, Migliore E, Camussi G et al. Post-apoptotic tumors are more palatable to dendritic cells and enhance their antigen cross-presentation activity. Vaccine 2008; 26: 6422–6432.
Janeway CA Jr, Medzhitov R . Innate immune recognition. Annu Rev Immunol 2002; 20: 197–216.
Matzinger P . Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045.
Lutz MB, Schuler G . Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol 2002; 23: 445–449.
Roux S, Apetoh L, Chalmin F, Ladoire S, Mignot G, Puig PE et al. CD4+CD25+ Tregs control the TRAIL-dependent cytotoxicity of tumor-infiltrating DCs in rodent models of colon cancer. J Clin Invest 2008; 118: 3751–3761.
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015; 520: 373–377.
Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, Toonen LW et al. Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int J Cancer 2008; 122: 1794–1802.
Vaught DB, Cook RS . Clearance of dying cells accelerates malignancy. Oncotarget 2015; 6: 24590–24591.
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med 2011; 208: 491–503.
Garg AD, De Ruysscher D, Agostinis P . Immunological metagene signatures derived from immunogenic cancer cell death associate with improved survival of patients with lung, breast or ovarian malignancies: a large-scale meta-analysis. Oncoimmunology 2015; doi: 10.1080/2162402X.2015.1069938.
Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2015; 136: E313–E325.
Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 2013; 210: 1685–1693.
Houot R, Kohrt HE, Marabelle A, Levy R . Targeting immune effector cells to promote antibody-induced cytotoxicity in cancer immunotherapy. Trends Immunol 2011; 32: 510–516.
Zhao T, Ren H, Wang X, Liu P, Yan F, Jiang W et al. Rituximab-induced HMGB1 release is associated with inhibition of STAT3 activity in human diffuse large B-cell lymphoma. Oncotarget 2015; 6: 27816–27831.
Cheadle EJ, Sidon L, Dovedi SJ, Melis MH, Alduaij W, Illidge TM et al. The induction of immunogenic cell death by type II anti-CD20 monoclonal antibodies has mechanistic differences compared with type I rituximab. Br J Haematol 2013; 162: 842–845.
Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV . FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell 2015; 28: 285–295.
Pardoll DM . The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264.
Mahoney KM, Rennert PD, Freeman GJ . Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 2015; 14: 561–584.
Sharma P, Allison JP . The future of immune checkpoint therapy. Science 2015; 348: 56–61.
Zappasodi R, de Braud F, Di Nicola M . Lymphoma immunotherapy: current status. Front Immunol 2015; 6: 448.
Bellocq JP, Luporsi E, Barriere J, Bonastre J, Chetritt J, Le Corroller AG et al. uPA/PAI-1, oncotype DX, MammaPrint((R)). Prognosis and predictive values for clinical utility in breast cancer management. Ann Pathol 2014; 34: 349–351.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6: 1–6.
Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One 2013; 8: e74250.
Gyorffy B, Surowiak P, Budczies J, Lanczky A . Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013; 8: e82241.
Garg AD, Maes H, van Vliet AR, Agostinis P . Targeting the hallmarks of cancer with therapy-induced endoplasmic reticulum (ER) stress. Mol Cell Oncol 2015; 2: e975089.
Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front Immunol 2015; 6: 402.
Van Gool S, Maes W, Ardon H, Verschuere T, Van Cauter S, De Vleeschouwer S . Dendritic cell therapy of high-grade gliomas. Brain Pathol 2009; 19: 694–712.
Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN . Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15: e257–e267.
Vandenberk L, Garg AD, Verschuere T, Koks C, Belmans J, Beullens M et al. Irradiation of necrotic cancer cells employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2015; doi: 10.1080/2162402X.2015.1083669.
Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M et al. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer Metastasis Rev 2011; 30: 61–69.
Ma Y, Conforti R, Aymeric L, Locher C, Kepp O, Kroemer G et al. How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev 2011; 30: 71–82.
Alexander HR Jr, Libutti SK, Pingpank JF, Steinberg SM, Bartlett DL, Helsabeck C et al. Hyperthermic isolated hepatic perfusion using melphalan for patients with ocular melanoma metastatic to liver. Clin Cancer Res 2003; 9: 6343–6349.
Taeger G, Grabellus F, Podleska LE, Muller S, Ruchholtz S . Effectiveness of regional chemotherapy with TNF-alpha/melphalan in advanced soft tissue sarcoma of the extremities. Int J Hyperthermia 2008; 24: 193–203.
Stach CM, Turnay X, Voll RE, Kern PM, Kolowos W, Beyer TD et al. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in Balb/c mice. Cell Death Differ 2000; 7: 911–915.
Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med 2015; 21: 1209–1215.
Vonderheide RH . CD47 blockade as another immune checkpoint therapy for cancer. Nat Med 2015; 21: 1122–1123.
Jinushi M, Sato M, Kanamoto A, Itoh A, Nagai S, Koyasu S et al. Milk fat globule epidermal growth factor-8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J Exp Med 2009; 206: 1317–1326.
Dudek AM, Garg AD, Krysko DV, De Ruysscher D, Agostinis P . Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev 2013; 24: 319–333.
Garg AD, Agostinis P . ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem Photobiol Sci 2014; 13: 474–487.
Korbelik M, Zhang W, Merchant S . Involvement of damage-associated molecular patterns in tumor response to photodynamic therapy: surface expression of calreticulin and high-mobility group box-1 release. Cancer Immunol Immunother 2011; 60: 1431–1437.
Korbelik M, Sun J, Cecic I . Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res 2005; 65: 1018–1026.
Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol 2015; 6: 187.
Wang Y, Yin C, Feng L, Wang C, Sheng G . Ara-C and anti-CD47 antibody combination therapy eliminates acute monocytic leukemia THP-1 cells in vivo and in vitro. Genet Mol Res 2015; 14: 5630–5641.
Panzarini E, Inguscio V, Fimia GM, Dini L . Rose Bengal acetate photodynamic therapy (RBAc-PDT) induces exposure and release of Damage-Associated Molecular Patterns (DAMPs) in human HeLa cells. PLoS One 2014; 9: e105778.
Shi Y, Fan X, Deng H, Brezski RJ, Rycyzyn M, Jordan RE et al. Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with Fcgamma receptors on macrophages. J Immunol 2015; 194: 4379–4386.
Pahl JH, Kwappenberg KM, Varypataki EM, Santos SJ, Kuijjer ML, Mohamed S et al. Macrophages inhibit human osteosarcoma cell growth after activation with the bacterial cell wall derivative liposomal muramyl tripeptide in combination with interferon-gamma. J Exp Clin Cancer Res 2014; 33: 27.
Tipton TR, Roghanian A, Oldham RJ, Carter MJ, Cox KL, Mockridge CI et al. Antigenic modulation limits the effector cell mechanisms employed by type I anti-CD20 monoclonal antibodies. Blood 2015; 125: 1901–1909.
Cheney CM, Stephens DM, Mo X, Rafiq S, Butchar J, Flynn JM et al. Ocaratuzumab, an Fc-engineered antibody demonstrates enhanced antibody-dependent cell-mediated cytotoxicity in chronic lymphocytic leukemia. mAbs 2014; 6: 749–755.
Antoine F, Ennaciri J, Girard D . Syk is a novel target of arsenic trioxide (ATO) and is involved in the toxic effect of ATO in human neutrophils. Toxicol In Vitro 2010; 24: 936–941.
Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 2015; 125: 1620–1636.
Wolf AM, Rumpold H, Tilg H, Gastl G, Gunsilius E, Wolf D . The effect of zoledronic acid on the function and differentiation of myeloid cells. Haematologica 2006; 91: 1165–1171.
Hyams JS, Donaldson MH, Metcalf JA, Root RK . Inhibition of human granulocyte function by methotrexate. Cancer Res 1978; 38: 650–655.
Sheu SJ, Chao YM, Liu NC, Chan JY . Differential effects of bevacizumab, ranibizumab and aflibercept on cell viability, phagocytosis and mitochondrial bioenergetics of retinal pigment epithelial cell. Acta Ophthalmol 2015; 93: e631–e643.
Mannerstrom M, Maenpaa H, Toimela T, Salminen L, Tahti H . The phagocytosis of rod outer segments is inhibited by selected drugs in retinal pigment epithelial cell cultures. Pharmacol Toxicol 2001; 88: 27–33.
Kovacs P, Csaba G . Effect of drugs affecting microtubular assembly on microtubules, phospholipid synthesis and physiological indices (signalling, growth, motility and phagocytosis) in Tetrahymena pyriformis. Cell Biochem Funct 2006; 24: 419–429.
Pallister CJ, Warnock DW . Effect of antimicrobial and antineoplastic drugs alone and in combination on the phagocytic and candidacidal function of human polymorphonuclear leucocytes. J Antimicrob Chemother 1989; 23: 87–94.
Klettner A, Tahmaz N, Dithmer M, Richert E, Roider J . Effects of aflibercept on primary RPE cells: toxicity, wound healing, uptake and phagocytosis. Br J Ophthalmol 2014; 98: 1448–1452.
Denes L, Szende B, Hajos G, Szporny L, Lapis K . Selective restoration of immunosuppressive effect of cytotoxic agents by thymopoietin fragments. Cancer Immunol Immunother 1990; 32: 51–54.
Medzihradsky JL, Wilson JG . Potentiation by thiopurines and sulfhydryl-reactive agents of the inhibition by 3-deazaadenosine of mononuclear phagocytes. Int J Immunopharmacol 1992; 14: 801–808.
Duong MN, Matera EL, Mathe D, Evesque A, Valsesia-Wittmann S, Clemenceau B et al. Effect of kinase inhibitors on the therapeutic properties of monoclonal antibodies. mAbs 2015; 7: 192–198.
Kumar V, Harjai K, Chhibber S . Thalidomide treatment modulates macrophage pro-inflammatory function and cytokine levels in Klebsiella pneumoniae B5055 induced pneumonia in BALB/c mice. Int Immunopharmacol 2010; 10: 777–783.
Lin JC, Liu CL, Lee JJ, Liu TP, Ko WC, Huang YC et al. Sorafenib induces autophagy and suppresses activation of human macrophage. Int Immunopharmacol 2013; 15: 333–339.
Lukac J, Kusic Z, Kordic D, Koncar M, Bolanca A . Natural killer cell activity, phagocytosis, and number of peripheral blood cells in breast cancer patients treated with tamoxifen. Breast Cancer Res Treat 1994; 29: 279–285.
Dong HP, Holth A, Kleinberg L, Ruud MG, Elstrand MB, Trope CG et al. Evaluation of cell surface expression of phosphatidylserine in ovarian carcinoma effusions using the annexin-V/7-AAD assay: clinical relevance and comparison with other apoptosis parameters. Am J Clin Pathol 2009; 132: 756–762.
Wemeau M, Kepp O, Tesniere A, Panaretakis T, Flament C, De Botton S et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis 2010; 1: e104.
Eric A, Juranic Z, Milovanovic Z, Markovic I, Inic M, Stanojevic-Bakic N et al. Effects of humoral immunity and calreticulin overexpression on postoperative course in breast cancer. Pathol Oncol Res 2009; 15: 89–90.
Peng RQ, Chen YB, Ding Y, Zhang R, Zhang X, Yu XJ et al. Expression of calreticulin is associated with infiltration of T-cells in stage IIIB colon cancer. World J Gastroenterol 2010; 16: 2428–2434.
Chen CN, Chang CC, Su TE, Hsu WM, Jeng YM, Ho MC et al. Identification of calreticulin as a prognosis marker and angiogenic regulator in human gastric cancer. Ann Surg Oncol 2009; 16: 524–533.
Liu R, Gong J, Chen J, Li Q, Song C, Zhang J et al. Calreticulin as a potential diagnostic biomarker for lung cancer. Cancer Immunol Immunother 2012; 61: 855–864.
Hsu WM, Hsieh FJ, Jeng YM, Kuo ML, Chen CN, Lai DM et al. Calreticulin expression in neuroblastoma—a novel independent prognostic factor. Ann Oncol 2005; 16: 314–321.
Baccelli I, Stenzinger A, Vogel V, Pfitzner BM, Klein C, Wallwiener M et al. Co-expression of MET and CD47 is a novel prognosticator for survival of luminal breast cancer patients. Oncotarget 2014; 5: 8147–8160.
Nagahara M, Mimori K, Kataoka A, Ishii H, Tanaka F, Nakagawa T et al. Correlated expression of CD47 and SIRPA in bone marrow and in peripheral blood predicts recurrence in breast cancer patients. Clin Cancer Res 2010; 16: 4625–4635.
Suzuki S, Yokobori T, Tanaka N, Sakai M, Sano A, Inose T et al. CD47 expression regulated by the miR-133a tumor suppressor is a novel prognostic marker in esophageal squamous cell carcinoma. Oncol Rep 2012; 28: 465–472.
Yoshida K, Tsujimoto H, Matsumura K, Kinoshita M, Takahata R, Matsumoto Y et al. CD47 is an adverse prognostic factor and a therapeutic target in gastric cancer. Cancer Med 2015; 4: 1322–1333.
Wang H, Tan M, Zhang S, Li X, Gao J, Zhang D et al. Expression and significance of CD44, CD47 and c-met in ovarian clear cell carcinoma. Int J Mol Sci 2015; 16: 3391–3404.
Chen JS, Hsu YM, Chen CC, Chen LL, Lee CC, Huang TS . Secreted heat shock protein 90alpha induces colorectal cancer cell invasion through CD91/LRP-1 and NF-kappaB-mediated integrin alphaV expression. J Biol Chem 2010; 285: 25458–25466.
Galluzzi L, Bravo-San Pedro JM, Kroemer G . Organelle-specific initiation of cell death. Nat Cell Biol 2014; 16: 728–736.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20: 1301–1309.
Acknowledgements
We thank Alexander R Van Vliet for critical reading of the manuscript. ADG is a recipient of the FWO postdoctoral fellowship 2013. NR is funded by European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 642295. This work was supported by grants from the Fund for Scientific Research Flanders (FWO-Vlaanderen; G.0661.09, G.0728.10 and G.0584.12N) and KU Leuven (GOA/11/009) to PA. This paper presents research results of the IAP7/32, funded by the Interuniversity Attraction Poles Programme, initiated by the Belgian State, Science Policy Office.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Kroemer
Rights and permissions
About this article

Cite this article
Garg, A., Romano, E., Rufo, N. et al. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: mechanisms and clinical translation. Cell Death Differ 23, 938–951 (2016). https://doi.org/10.1038/cdd.2016.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.5
This article is cited by
-
The GAR/RGG motif defines a family of nuclear alarmins
Cell Death & Disease (2021)
-
The bovine dialysable leukocyte extract IMMUNEPOTENT CRP induces immunogenic cell death in breast cancer cells leading to long-term antitumour memory
British Journal of Cancer (2021)
-
Impact of aging on gene expression response to x-ray irradiation using mouse blood
Scientific Reports (2021)
-
Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer
Cell Death & Disease (2020)
-
ASC-J9® increases the bladder cancer chemotherapy efficacy via altering the androgen receptor (AR) and NF-κB survival signals
Journal of Experimental & Clinical Cancer Research (2019)
