
Abstract
Autophagy is a homeostatic process that enables eukaryotic cells to deliver cytoplasmic constituents for lysosomal degradation, to recycle nutrients and to survive during starvation. In addition to these primordial functions, autophagy has emerged as a key mechanism in orchestrating innate and adaptive immune responses to intracellular pathogens. Autophagy restricts viral infections as well as replication of intracellular bacteria and parasites and delivers pathogenic determinants for TLR stimulation and for MHC class II presentation to the adaptive immune system. Apart from its role in defense against pathogens, autophagy-mediated presentation of self-antigens in the steady state could have a crucial role in the induction and maintenance of CD4+ T-cell tolerance. This review describes the mechanisms by which the immune system utilizes autophagic degradation of cytoplasmic material to regulate adaptive immune responses.
Similar content being viewed by others


Main
Eukaryotic cells contain two major protein degradation systems, proteasomes and lysosomes. For proteasomes, polyubiquitination allows substrates to gain access to the catalytic chamber for proteolysis.1 Lysosomal degradation of exogenous material is mediated by the process of endocytosis/phagocytosis, whereas degradation of cytoplasmic components occurs after autophagy. How autophagosome cargo is selected remains unclear,2 but in contrast to proteasomes, which degrade soluble short-lived proteins, autophagy targets cell organelles and aggregates of long-lived proteins for degradation.3 Basal autophagy is ubiquitous in eukaryotes. It allows recycling of nutrients and removal of damaged or unwanted cytosolic proteins and organelles.4
Apart from its functions in preserving cellular bioenergetics, autophagy appears to have a protective role against diverse pathologies owing to its cellular clearance function and because of removal of damaged or aggregate-prone proteins.5, 6, 7, 8 In keeping with the clearance function, autophagy assists in the restriction and elimination of intracellular pathogens as an innate immune response to viral and microbial infection. In addition, by delivering cytoplasmic antigens for loading onto major histocompatibility complex (MHC) class II molecules for CD4+ T-cell recognition, autophagy enables the immune surveillance for intracellular antigens and broadens the immunological functions of MHC class II presentation. Autophagy plays a role in the survival and cell death of adaptive immune cells and is, in turn, regulated by innate and adaptive immune signals. Here, we provide an introduction to autophagy as an innate immune defense mechanism and highlight its role in the initiation and execution of adaptive immune responses.
Autophagy Pathways
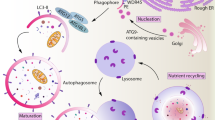
There are at least three distinct pathways of autophagy: microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy. Microautophagy is characterized by the uptake of cytoplasmic components at the lysosomal membrane via budding into the lysosome, through poorly defined mechanism. During CMA, proteins are directly imported into lysosomes through the LAMP-2a transporter9, 10 assisted by cytosolic and lysosomal HSC70 chaperones (Figure 1). Substrates of chaperone-mediated autophagy carry signal peptides for sorting into lysosomes, similar to other protein transport mechanisms across membranes.11

Cellular and molecular events during macroautophagy and chaperone-mediated autophagy. The autophagosome is formed by expansion of a membrane core of unknown origin, termed the phagophore or isolation membrane and is initiated by a complex of the type III PI3K and Atg6 (Beclin-1). Two ubiquitin-like systems are required for elongation of the isolation membrane: the Atg8/LC3 and the Atg12 conjugation system. The five C-terminal amino acids of Atg8/LC3 are cleaved of by Atg4 to reveal glycine 120 (G120), which is required to link the protein after activation by Atg7 and ligation by Atg3 to PE in the autophagosomal membrane (green circles). Similarly, glycine 140 (G140) is used by Atg7 and Atg10 to couple Atg12 with Atg5. This complex then localizes to the outer membrane of the forming autophagosome (blue squares). Upon autophagosome completion, the Atg12–Atg5 complex recycles from the outer membrane, and only Atg8 (LC3) remains associated with the completed autophagosome. Autophagosomes then fuse with late endosomes and lysosomes for degradation of their cargo and their intravesicular membranes. During CMA, proteins are directly imported into lysosomes through the LAMP-2a transporter assisted by cytosolic and lysosomal HSC70 chaperones
Macroautophagy is the major route of degradation of cytoplasmic constituents. During macroautophagy, cytosolic constituents including organelles are enclosed in a double-membrane vesicle, called autophagosome,12, 13 which then fuses with lysosomes and late endosomes for degradation of the inner autophagosomal membrane and its cargo14 (Figure 1). The resulting breakdown products of macromolecules are subsequently released back into the cytosol through permeases in the lysosomal membrane, where they can be reused for anabolic or catabolic reactions.15
The execution of macroautophagy is mediated by evolutionary conserved proteins known as autophagy-related (Atg) proteins. Yeast genetic studies have identified more than 30 Atg genes required for macroautophagy.16 Atg6 (also known as Beclin-1) and its binding partner, the class III phosphatidylinositol 3-kinase (PI3K), are required for the initiation of the isolation membrane. The elongation and shape of the autophagosome are regulated by at least two ubiquitin-like systems: the Atg8 (with its mammalian homologues LC3, GATE-16, GABARAP and Apg8L) and the Atg12 conjugation pathways (Figure 1). The five C-terminal amino acids of Atg8/LC3 are cleaved off by the Atg4 protease to liberate a glycine residue (G120). This C-terminal residue then gets transferred to phosphatidylethanolamine (PE) in the forming isolation membrane by the E1- and E2-like enzymes Atg7 and Atg3. Although LC3/Atg8 gets recycled from the outer autophagosomal membrane by deconjugation from its phospholipid, it remains attached to the inner autophagosomal membrane, and this portion is degraded with the inner autophagosomal membrane in lysosomes and late endosomes after fusion with these vesicles.17 The more autophagosomes are formed, the more LC3 is degraded in autolysosomes, and therefore, lysosomal turnover of LC3 can be used as a measure for macroautophagic activity.18 In addition, autophagosome-associated LC3 (called LC3-II) and free cytosolic LC3 (called LC3-I) can be distinguished by their apparent molecular weights in SDS-PAGE gel electrophoresis (16 and 18 kDa, respectively) and thus can be quantified separately in anti-LC3 immunoblots.17, 19 Therefore, LC3-II turnover has become a universal measurement for the autophagic flux, and autophagosomes can be visualized with Atg8/LC3, coupled with fluorescent proteins.20
In the other ubiquitin-like system, Atg12 gets coupled through its C-terminal glycine residue (G140) to a lysine residue of Atg5 by the E1- and E2-like enzymes Atg7 and Atg10. The Atg12–Atg5 complex associates with Atg16 and then binds to the outer surface of the isolation membrane. Upon completion of the autophagosome, the Atg5–Atg12–Atg16 complex dissociates from the outer autophagosomal membrane and only Atg8/LC3 remains associated with the completed autophagosome. Autophagosomes then fuse with late endosomes and lysosomes for degradation of their cargo and their intravesicular membranes.
Studies on the molecular mechanisms of macroautophagy and its importance in protein metabolism have set the stage to analyze its role in multiple biological processes including innate and adaptive immune responses. Macroautophagy is now recognized to restrict viral infections and replication of intracellular bacteria and parasites. In addition, this pathway delivers cytoplasmic antigens for MHC class II presentation to the adaptive immune system, which then in turn is able to regulate macroautophagy. At the same time, macroautophagy plays a role in T-cell survival and effector function. Thus, the immune system utilizes macroautophagy, to both restrict intracellular pathogens and to regulate innate and adaptive immunity.
Autophagy in Innate and Adaptive Immunity
As an innate immune response to viral and microbial pathogens, macroautophagy participates in limiting pathogen replication in host cells by at least three mechanisms: (i) free bacteria, such as group A Streptococci, become enveloped by autophagosomes and are delivered for lysosomal hydrolysis.21 (ii) Macroautophagy targets phagosomes that have become conditioned by bacteria or parasites to avoid fusion with lysosomes, such as in Mycobacterium (M) tuberculosis infection.22, 23 (iii) Finally, delivery of cytoplasmic pathogen-associated molecular patterns (PAMPs) such as viral nucleic acids to endosomal toll-like receptors (TLR) through macroautophagy results in robust type I IFN-dependent innate immune responses24 (Figure 2).

Autophagy in innate and adaptive immunity. Autophagy orchestrates innate and adaptive immune responses to intracellular pathogens and is regulated by innate and adaptive immune signals. Intracellular pathogens engulfed into autophagosomes are degraded in autolysosomes. Viral nucleic acids (ssRNA) can be transferred by autophagosomes to TLR containing endosomes which signal the induction of type I interferon responses and enhance macroautophagy. Similarly, extracellular pathogen patterns can stimulate macroautophagy through binding to surface TLRs. In addition, autophagic pathways can deliver antigens for MHC class II presentation. Autophagosomes and LAMP-2a, the transporter associated with chaperone-mediated autophagy, can transport antigens into the MHC class II containing compartment (MIIC). In MIICs, the antigen is processed and loaded onto MHC class II molecules for CD4+ T-cell stimulation. Activated CD4+ T cells can then in turn enhance macroautophagy and autophagosome-lysosome fusion through type II IFN and TNF family members (IFN-γ, TNF, TRAIL, and CD40L)
Notably, several prototype TLR ligands such as poly(I:C) (TLR3), LPS (TLR4) and single-stranded RNA (TLR7), are capable of inducing macroautophagy in a murine macrophage cell line (RAW 264.7).25, 26 In the latter study, stimulation of macroautophagy through TLR7 resulted in elimination of phagocytosed vaccine strain M. bovis bacille Calmette-Guérin (BCG). Thus, macroautophagy does not only target PAMPs to endosomal TLRs for immune recognition, but is also susceptible to TLR stimulation suggesting that the TLR signaling pathway could be exploited for the elimination of intracellular pathogens through induction of macroautophagy.26
In addition to TLR signaling, cytokines such as interferons (IFNs) and members of the tumor necrosis factor (TNF) receptor ligand family such as CD40-CD40L stimulation are capable of modulating macroautophagy (Figure 2). Both types I and II IFNs have been reported to induce macroautophagy in susceptible cells. Restriction of HSV-1 infection by macroautophagy in vitro and in vivo was found to be dependent on type I IFN signaling machinery.27, 28 Type II IFN has been reported to enhance M. tuberculosis and Ricksettia conorii degradation by macroautophagy in infected cells.22, 23 IFN-γ induces macroautophagy and mycobacterial clearance through immunity-related GTPases (IRGs).29 Mouse tissues are probably more susceptible to this macroautophagy regulation mechanism because their IRGs are IFN-γ inducible, whereas the human IRG is not, suggesting that immune signals that stimulate macroautophagy differ between rodents and man.
TNF-α was found to upregulate macroautophagy in cells lacking NF-κB activation30 and TNF-related apoptosis-inducing ligand (TRAIL) was described to induce macroautophagy in the human epithelial cells.31 Consistent with this, inactivation of Fas-associated death domain, the signaling adapter protein of the TRAIL receptor, decreases macroautophagy induction by TRAIL.32 As a third TNF family member, CD40L has been demonstrated to induce macroautophagy-mediated fusion of Toxoplasma gondii-containing phagosomes with lysosomes through CD40 signaling on mouse and human macrophages.33
In addition to limiting pathogen replication in host cells, macroautophagy also delivers viral, parasitic, and bacterial antigens to late endosomal compartments, where macroautophagy substrates are then degraded by lysosomal hydrolases. The fusion vesicles between autophagosomes and late endosomes, the so-called amphisomes, display a multivesicular and multilamellar morphology reminiscent of MHC class II containing compartments (MIICs).34
Indeed, a number of studies provide evidence that nuclear and cytosolic antigens are delivered onto MHC class II molecules through macroautophagy or chaperone-mediated autophagy.19, 35, 36, 37, 38, 39, 40 In this context, we could previously show that GFP-Atg8 (GFP-LC3)-positive autophagosomes fuse frequently with MIICs, as identified by the presence of MHC class II, the lysosomal membrane protein LAMP-2 and the chaperone HLA-DM, which is involved in peptide loading onto MHC class II molecules.19 Targeting a model antigen into this pathway through fusion of the autophagosome marker LC3 resulted in a strong increase in MHC class II presentation and CD4+ T-cell recognition19 indicating that macroautophagy efficiently delivers intracellular antigens for MHC class II presentation and CD4+ T-cell recognition.
These data indicate that the relationship between macroautophagy and immunity is bidirectional and that the immune system utilizes macroautophagy to both restrict intracellular pathogens and to regulate adaptive immunity (Figure 2). At the same time, the efficacy and sustainability of adaptive immune responses appear to be determined by the capacity to perform macroautophagy, as discussed in the next paragraph.
Role of Autophagy in T- and B-Cell Function and Survival
Autophagy has a central but complex role in cell survival in numerous cell types, functioning either as a pro-survival or as a cell death mechanism depending on the cell type, the nature of the death stimulus and subsequent compensatory changes.41, 42 These divergent functions on cell survival are also reflected in the role of macroautophagy for T-cell survival (Figure 3). By using lethally irradiated mice repopulated with haematopoetic cells from fetal livers of atg5−/− mice, Pua et al.43 showed that atg5−/− CD4+ and CD8+ T cells developed normally in the recipient thymus, but failed to repopulate the periphery because of massive cell death (predominantly of CD8+ T cells) and failed to undergo efficient proliferation after T-cell receptor stimulation. These data indicate a critical role for Atg5/macroautophagy in lymphocyte development and function and suggest that macroautophagy may be essential for both T lymphocyte survival and proliferation in the steady state and after immune activation. Future studies will show whether and to what degree these results can be extrapolated to other lymphoid and myeloid cells of the immune system, such as innate lymphocytes and dendritic cells (DCs). Furthermore, it is currently not known whether distinct functional subsets of lymphocytes, for example, effector versus memory T cells, are particularly dependent on macroautophagy.

Autophagy in lymphocyte survival. Autophagy has a complex role on cell survival in immune cells, functioning either as pro-survival or as cell death mechanism. Highly proliferative lymphocyte compartments of the adaptive immune system such as T cells and B cells appear to require macroautophagy to efficiently mobilize nutrients and maintain cellular fitness both during development and immune responses (a), whereas Th2-polarized CD4+ T cells are susceptible to cell death by macroautophagy (b)
In contrast to its function as a T-cell survival mechanism, Li et al.44 reported that Th2-polarized CD4+ T cells, which are thought to support humoral immune responses most efficiently, display more GFP-Atg8/LC3+ autophagosomes than Th1 cells, which are particularly important for cell-mediated immune responses. Th2 cells were found to be more resistant to cell death upon growth factor withdrawal when macroautophagy was inhibited upon RNA silencing of Atg7 or Atg6 (Beclin-1), whereas steady-state survival and proliferation were not affected by macroautophagy inhibition. These data indicate that Th2-polarized CD4+ T cells, which show a high level of autophagic activity, utilize the macroautophagy machinery to execute apoptotic cell death. In line with this interpretation, HIV envelope glycoproteins have been shown to induce autophagic cell death by binding to CXC-chemokine receptor 4 on uninfected bystander CD4+ T cells, which could be blocked by RNA silencing of Atg7 or Atg6 (Beclin-1).45
Similar to T-cell development, B cells require macroautophagy both during development and maintenance in the periphery. Atg5-deficient pro-B cells do not efficiently develop into pre-B cells, but instead, seem to die at increased frequencies of apoptosis.46 This leads to reduced B-1 cell numbers in the periphery. In addition, B-1a cell survival is compromised in the absence of Atg5. This suggests that highly proliferative lymphocyte compartments of the adaptive immune system require macroautophagy to efficiently mobilize nutrients and maintain cellular fitness both during development and immune responses.
Antigen Processing for MHC Presentation
As this review focuses on the role of autophagy in mediating CD4+ T-cell responses and in regulating CD4+ T-cell immunity through processing and presentation of intracellular antigens on MHC class II molecules, we will briefly discuss the classical paradigm of antigen processing for MHC presentation. The two main classes of classical and polymorphic MHC molecules are loaded with protein fragments in distinct cellular compartments and their peptide cargo reaches these compartments by different routes. Antigens for MHC class I presentation are derived from proteins that are degraded in the cytosol by the proteasome, a large cytosolic enzyme complex. Targeting these antigens for proteasomal degradation is often mediated by ubiquitinylation. A significant proportion of these peptides are thought to be generated from defective ribosomal products, which are newly synthesized proteins that fail to fold properly or whose translation was terminated prematurely. To fit into the closed groove of the MHC class I molecule, the peptides generated by the proteasome, many of which are more than 10 amino acids in length, undergo further trimming by aminopeptidases in the cytosol and in the endoplasmic reticulum. With the help of the MHC class I loading complex, which includes chaperones, aminopeptidases and thiol oxidoreductases individual peptides of 8–9 amino acids in length are loaded into the peptide-binding groove of MHC class I molecules. Stable peptide–MHC class I complexes are then exported through the Golgi apparatus to the cell surface for recognition by CD8+ T cells.
MHC class II molecules are constitutively expressed on phagocytes, dendritic cells, B cells, lymph node stromal cells and thymic epithelial cells where they participate in the selection of the T-cell receptor repertoire. In addition, cytokines such as IFN-γ can induce MHC class II surface expression on non-professional antigen-presenting cells, and activation induces MHC class II on nearly all human lymphocytes. MHC class II molecules bind peptides generated in the endosomal--lysosomal systems and display them on the cell surface for recognition of CD4+ T cells. Antigens reach this compartment often by endocytosis and are then degraded by lysosomal endo- and exoproteases. They meet MHC class II molecules in the so-called MIICs or class II vesicles as outlined above. MHC class II peptides are generated by a number of lysosomal proteases, which together with the open-ended peptide-binding cleft of MHC class II molecules are thought to enable the MHC molecules to capture a maximal range of potential antigenic peptides. Like classical MHC class I molecules, newly synthesized MHC class II molecules are unstable in the absence of bound peptide. Therefore, the transmembrane protein invariant chain (Ii) protein blocks the peptide-binding groove of newly synthesized MHC class II molecules to prevent premature binding of antigens. In addition, the li contains an endosomal targeting signal and thus targets MHC class II molecules to late endosomes, where they meet peptides generated by lysosomal proteases. In this MHC class II loading compartment, lysosomal proteases also degrade the Ii, and the remaining peptide (CLIP for class II-associated Ii peptide) is exchanged for antigenic peptides with the help of the non-classical MHC class II molecule HLA-DM. As a result of this pathway, MHC class II ligands are generated from extracellular antigens after endocytosis and degradation in lysosomes.
The finding that professional APCs, especially DCs, are able to present extracellular antigen not only on MHC class II, but also on MHC class I through cross-presentation challenged the traditional paradigm that MHC class I antigens are exclusively of cytosolic or nuclear origin. Cross-presentation allows DCs to prime CD8+ T-cell responses to antigens synthesized by cells other than DCs and to trigger both CD8+ and CD4+ T-cell responses at the same time, generating more effective and sustained T-cell responses. Similarly, pathogens that replicate in the cytoplasm of antigen-presenting cells should be detectable for the immune system through both MHC class I and MHC class II presentation.
Autophagy and Antigen Presentation
As cross-presentation of extracellular antigen onto MHC class I molecules changed our understanding of antigen processing for MHC class I presentation, several lines of evidence support the notion that intracellular antigen can also access MHC class II presentation (Table 1). The first evidence for the existence of an endogenous MHC class II pathway came from studies on viral antigen presentation to CD4+ T cells. Eric Long and coworkers47, 48 described that measles and influenza antigens could be presented to CD4+ T cells through an intracellular route that did not sensitize bystander cells for recognition. Additional support for this alternate MHC class II loading pathway was found in the analysis of natural MHC class II ligands. When MHC class II molecules were affinity-purified, primarily from Epstein–Barr virus (EBV)-transformed B lymphoblastoid cell lines, murine B cell lymphoma and human as well as mouse myeloid cells, the majority of natural MHC class II ligands was found to be derived from intracellular proteins.65 Although the majority of natural MHC class II ligands is derived from membrane and secreted proteins, more than 20% originate from cytosolic and nuclear antigens.36, 55, 66 A prominent source of human MHC class II ligands is the glyceraldehyde-3-phosphate dehydrogenase, which gives rise to natural peptide ligands for five different HLA class II alleles, but has not been eluted from MHC class I molecules so far.36, 67 This prominent protein of MHC class II presentation was shown to be degraded via chaperone-mediated autophagy68 and could be isolated from autophagosomes.12 Moreover, peptides of two mammalian Atg8 homologues, LC3 and GABARAP, have been isolated from human and mouse MHC class II molecules,36, 56 and the latter was even recognized by a T-cell clone isolated from the pancreas draining lymph node of a NOD mouse. These data suggest that macroautophagy and chaperone-mediated autophagy could be involved in intracellular antigen processing for MHC class II loading.
This suspicion was indeed confirmed initially by pharmacological inhibition of macroautophagy with 3-methyladenine (3-MA). Overexpression of complement C5,35 neomycin phosphotransferase II38 and mucin 137 led to intracellular antigen processing onto MHC class II, which was 3-MA sensitive. With more specific inhibition of macroautophagy, namely targeting of Atg12 by siRNA-mediated gene silencing, we could show that the nuclear antigen 1 of EBV (EBNA1), which is intracellularly processed onto MHC class II,69 requires macroautophagy to be presented on EBV-transformed B cells to CD4+ T cells.39 In parallel, overexpression of the molecular machinery of chaperone-mediated autophagy was shown to enhance MHC class II presentation of two autoantigens, glutamate decarbozylase 65 (GAD65) and the mutant human immunoglobulin κ chain SMA.40 Both LAMP-2a and HSC70 overexpression enhanced presentation of these two autoantigens to CD4+ T cells. Therefore, both macroautophagy and chaperone-mediated autophagy seem to be involved in intracellular processing of some antigens for MHC class II presentation to CD4+ T cells. Further studies will have to analyze to what extent this pathway contributes to priming and tolerance of CD4+ T cells, as well as immune surveillance by these adaptive lymphocytes in vivo.
Autophagy in Tolerance and Autoimmunity
The function of macroautophagy and chaperone-mediated autophagy in facilitating cytoplasmic self- or foreign-antigen recognition by CD4+ T cells has primarily been studied in cell culture models. Thus, the in vivo significance of these pathways remains yet to be clearly established. However, recent studies suggest that macroautophagy could play an important role in maintaining T-cell tolerance and that defects in the macroautophagy pathway might contribute to the development of autoimmune diseases.
The chief mechanism of T-cell tolerance is the deletion of self-reactive T cells in the thymus, a process called central tolerance induction.70 Cells whose T-cell receptors have a too low affinity for peptides derived from endogenous proteins bound to MHC molecules do not receive a signal to switch off the process of spontaneous apoptosis and therefore die in the thymus. Cells whose T-cell receptors have a high affinity for such complexes presented by medullary thymic epithelial cells and bone marrow-derived thymic dendritic cells are also eliminated by means of apoptosis (negative selection) or are converted into a regulatory cell type following induction of the FoxP3 transcriptional regulator. Some self-reactive lymphocytes exit the thymus and enter the periphery as mature T cells, and this necessitates the existence of peripheral mechanisms that participate in T-cell tolerance. One of these mechanisms is deletion or abortive activation of self-reactive lymphocytes by autoantigen-presenting cells that express low levels of costimulators.71
Direct self-antigen presentation on MHC class II through macroautophagy in thymic epithelial cells as well as thymic and peripheral professional and non-professional antigen-presenting cells72 could contribute to central and peripheral tolerance maintenance in the CD4+ T-cell compartment. In agreement with this hypothesis, thymic epithelia display great amounts of autophagosomes in the GFP-LC3 transgenic macroautophagy reporter mouse in the steady state.73 Notably, more autophagosomes were found in this tissue in newborn compared with adult mice. These findings correlate with T-cell selection and central tolerance induction for CD4+ T cells being most active at a young age.70 In addition, we find considerable macroautophagy in immature dendritic cells,19 which have been implicated in peripheral tolerance induction.71
Some of these tolerance mechanisms might be compromised in patients with mutations in autophagy genes, resulting in Crohn's disease, a chronic immune-mediated bowel disease. Several recent genome-wide scans identified a single-nucleotide polymorphism in the autophagy gene ATG16L1 (T300A variant),74, 75, 76 involved in autophagosome formation, as well as in the macroautophagy-stimulatory GTPase IRGM77 as novel risk-conferring genes in Crohn's disease. Although it is not established whether these mutations lead to a defective function of macroautophagy, it is tempting to speculate that decreased macroautophagy because of Atg16 mutations might either impair innate resistance to invading bacteria in intestinal epithelial cells and thereby trigger inflammation as a result of increased antigenic load or lead to insufficient tolerance induction against commensals or self-antigens in the gut.
Another possible link between macroautophagy, tolerance and autoimmunity is the role of macroautophagy in the removal of apoptotic cell corpses.78 Rapid removal of apoptotic cell material is thought to be crucial for the prevention of tissue inflammation. Qu et al.78 found that dying cells in macroautophagy-deficient atg5−/− embryos fail to express signals that ensure their clearance. This functional deficit was associated with increased inflammation in tissues that showed impaired clearance of apoptotic cells. Defective clearance of apoptotic cells has long been suggested to drive autoimmunity in patients with systemic lupus erythematosus and other autoimmune diseases.79
Thus, macroautophagy potentially prevents inflammatory tissue damage and the generation of autoimmune effector responses owing to sensitization of dying cells for early recognition and non-inflammatory removal. In addition, macroautophagy might be crucial in the induction and maintenance of CD4+ T-cell tolerance through thymic T-cell repertoire selection and by mediating peripheral CD4+ T-cell tolerance toward self-antigens.
Conclusion
Previous studies provided evidence that autophagy plays an important role in eliminating intracellular pathogens and in mediating CD4+ T-cell recognition of endogenous antigens, although the substrate requirements for pathogen and antigen import into autophagsomes remain to be defined. In turn, innate and adaptive immune signals can induce, augment or inhibit autophagy in susceptible cell types. Moreover, autophagy might be essential in maintaining cellular fitness of innate and adaptive immune effector cells during immune responses. A role for autophagy in central and peripheral T-cell tolerance and in the prevention of autoimmune diseases is intriguing but remains speculative. In vivo studies in conditional knockout mice for essential autophagy genes should provide a better understanding of how and by which mechanisms autophagy regulates CD4+ T-cell immunity and tolerance in the steady state and during immune activation.
Abbreviations
- CMA:
-
chaperone-mediated autophagy
- Atg:
-
autophagy-related gene
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- MHC:
-
major histocompatibility complex
- DRiPs:
-
defective ribosomal products
- TAP:
-
transporter associated with antigen processing
- Ii:
-
invariant chain
- CLIP:
-
class II-associated Ii peptide
- APC:
-
antigen-presenting cell
- MIIC:
-
MHC class II containing compartment
- HLA:
-
human leukocyte antigen
- DC:
-
dendritic cell
- EBV:
-
Epstein–Barr virus
- LAMP:
-
lysosome-associated membrane protein
References
Ciechanover A, Finley D, Varshavsky A . Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 1984; 37: 57–66.
De Duve C, Wattiaux R . Functions of lysosomes. Annu Rev Physiol 1966; 28: 435–492.
Henell F, Berkenstam A, Ahlberg J, Glaumann H . Degradation of short- and long-lived proteins in perfused liver and in isolated autophagic vacuoles – lysosomes. Exp Mol Pathol 1987; 46: 1–14.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–884.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–889.
Lünemann JD, Schmidt J, Schmid D, Barthel K, Wrede A, Dalakas MC et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann Neurol 2007; 61: 476–483.
Martinez-Vicente M, Cuervo AM . Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 2007; 6: 352–361.
Cuervo AM, Dice JF . A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996; 273: 501–503.
Cuervo AM, Dice JF . Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci 2000; 113 (Part 24): 4441–4450.
Agarraberes FA, Dice JF . A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 2001; 114 (Part 13): 2491–2499.
Fengsrud M, Erichsen ES, Berg TO, Raiborg C, Seglen PO . Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur J Cell Biol 2000; 79: 871–882.
Stromhaug PE, Berg TO, Fengsrud M, Seglen PO . Purification and characterization of autophagosomes from rat hepatocytes. Biochem J 1998; 335 (Part 2): 217–224.
Ohsumi Y . Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2: 211–216.
Mizushima N, Klionsky DJ . Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 2007; 27: 19–40.
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E . Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1: 84–91.
Schmid D, Pypaert M, Münz C . Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007; 26: 79–92.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4: 151–175.
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T et al. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306: 1037–1040.
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V . Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119: 753–766.
Singh SB, Davis AS, Taylor GA, Deretic V . Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313: 1438–1441.
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A . Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007; 315: 1398–1401.
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT . Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27: 135–144.
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V . Toll-like receptors control autophagy. EMBO J 2008; 27: 1110–1121.
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007; 1: 23–35.
Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 2002; 99: 190–195.
Taylor GA, Feng CG, Sher A . p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol 2004; 4: 100–109.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281: 30373–30382.
Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS . Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci USA 2004; 101: 3438–3443.
Thorburn J, Moore F, Rao A, Barclay WW, Thomas LR, Grant KW et al. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol Biol Cell 2005; 16: 1189–1199.
Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS . CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest 2006; 116: 2366–2377.
Zwart W, Griekspoor A, Kuijl C, Marsman M, van Rheenen J, Janssen H et al. Spatial separation of HLA-DM/HLA-DR interactions within MIIC and phagosome-induced immune escape. Immunity 2005; 22: 221–233.
Brazil MI, Weiss S, Stockinger B . Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur J Immunol 1997; 27: 1506–1514.
Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 2005; 102: 7922–7927.
Dorfel D, Appel S, Grunebach F, Weck MM, Muller MR, Heine A et al. Processing and presentation of HLA class I and II epitopes by dendritic cells after transfection with in vitro-transcribed MUC1 RNA. Blood 2005; 105: 3199–3205.
Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW et al. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol 2003; 33: 1250–1259.
Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307: 593–596.
Zhou D, Li P, Lin Y, Lott JM, Hislop AD, Canaday DH et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 2005; 22: 571–581.
Kroemer G, Jaattela M . Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5: 886–897.
Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem 2008; 283: 4766–4777.
Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW . A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204: 25–31.
Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol 2006; 177: 5163–5168.
Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 2006; 116: 2161–2172.
Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008; 4: 309–314.
Jacobson S, Sekaly RP, Jacobson CL, McFarland HF, Long EO . HLA class II-restricted presentation of cytoplasmic measles virus antigens to cytotoxic T cells. J Virol 1989; 63: 1756–1762.
Jaraquemada D, Marti M, Long EO . An endogenous processing pathway in vaccinia virus-infected cells for presentation of cytoplasmic antigens to class II-restricted T cells. J Exp Med 1990; 72: 947–954.
Nuchtern JG, Biddison WE, Klausner RD . Class II MHC molecules can use the endogenous pathway of antigen presentation. Nature 1990; 343: 74–76.
Gueguen M, Long EO . Presentation of a cytosolic antigen by major histocompatibility complex class II molecules requires a long-lived form of the antigen. Proc Natl Acad Sci USA 1996; 93: 14692–14697.
Aichinger G, Karlsson L, Jackson MR, Vestberg M, Vaughan JH, Teyton L et al. Major histocompatibility complex class II-dependent unfolding, transport, and degradation of endogenous proteins. J Biol Chem 1997; 272: 29127–29136.
Malnati MS, Marti M, LaVaute T, Jaraquemada D, Biddison W, DeMars R et al. Processing pathways for presentation of cytosolic antigen to MHC class II-restricted T cells. Nature 1992; 357: 702–704.
Chen M, Shirai M, Liu Z, Arichi T, Takahashi H, Nishioka M . Efficient class II major histocompatibility complex presentation of endogenously synthesized hepatitis C virus core protein by Epstein-Barr virus-transformed B-lymphoblastoid cell lines to CD4+ T cells. J Virol 1998; 72: 8301–8308.
Lich JD, Elliott JF, Blum JS . Cytoplasmic processing is a prerequisite for presentation of an endogenous antigen by major histocompatibility complex class II proteins. J Exp Med 2000; 191: 1513–1524.
Dongre AR, Kovats S, deRoos P, McCormack AL, Nakagawa T, Paharkova-Vatchkova V et al. In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur J Immunol 2001; 31: 1485–1494.
Suri A, Walters JJ, Rohrs HW, Gross ML, Unanue ER . First signature of islet beta-cell-derived naturally processed peptides selected by diabetogenic class II MHC molecules. J Immunol 2008; 180: 3849–3856.
Bonifaz LC, Arzate S, Moreno J . Endogenous and exogenous forms of the same antigen are processed from different pools to bind MHC class II molecules in endocytic compartments. Eur J Immunol 1999; 29: 119–131.
Qi L, Rojas JM, Ostrand-Rosenberg S . Tumor cells present MHC class II-restricted nuclear and mitochondrial antigens and are the predominant antigen presenting cells in vivo. J Immunol 2000; 165: 5451–5461.
Mukherjee P, Dani A, Bhatia S, Singh N, Rudensky AY, George A et al. Efficient presentation of both cytosolic and endogenous transmembrane protein antigens on MHC class II is dependent on cytoplasmic proteolysis. J Immunol 2001; 167: 2632–2641.
Oukka M, Cohen-Tannoudji M, Tanaka Y, Babinet C, Kosmatopoulos K . Medullary thymic epithelial cells induce tolerance to intracellular proteins. J Immunol 1996; 156: 968–975.
Oukka M, Colucci-Guyon E, Tran PL, Cohen-Tannoudji M, Babinet C, Lotteau V et al. CD4 T cell tolerance to nuclear proteins induced by medullary thymic epithelium. Immunity 1996; 4: 545–553.
Dani A, Chaudhry A, Mukherjee P, Rajagopal D, Bhatia S, George A et al. The pathway for MHCII-mediated presentation of endogenous proteins involves peptide transport to the endo-lysosomal compartment. J Cell Sci 2004; 117 (Part 18): 4219–4230.
Weiss S, Bogen B . MHC class II-restricted presentation of intracellular antigen. Cell 1991; 64: 767–776.
Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA . Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science 1999; 284: 1351–1354.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S . SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999; 50: 213–219.
Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL . Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med 1993; 178: 27–47.
Friede T, Gnau V, Jung G, Keilholz W, Stevanovic S, Rammensee HG . Natural ligand motifs of closely related HLA-DR4 molecules predict features of rheumatoid arthritis associated peptides. Biochim Biophys Acta 1996; 1316: 85–101.
Aniento F, Roche E, Cuervo AM, Knecht E . Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J Biol Chem 1993; 268: 10463–10470.
Münz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG et al. Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J Exp Med 2000; 191: 1649–1660.
Starr TK, Jameson SC, Hogquist KA . Positive and negative selection of T cells. Annu Rev Immunol 2003; 21: 139–176.
Steinman RM, Hawiger D, Nussenzweig MC . Tolerogenic dendritic cells. Annu Rev Immunol 2003; 21: 685–711.
Lee JW, Epardaud M, Sun J, Becker JE, Cheng AC, Yonekura AR et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol 2007; 8: 181–190.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y . In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–1111.
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39: 207–211.
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A et al. Genome-wide association study identifies new susceptibility loci for Crohn's disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39: 596–604.
Prescott NJ, Fisher SA, Franke A, Hampe J, Onnie CM, Soars D et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn's disease and is independent of CARD15 and IBD5. Gastroenterology 2007; 132: 1665–1671.
Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 2007; 39: 830–832.
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931–946.
Bratton DL, Henson PM . Autoimmunity and apoptosis: refusing to go quietly. Nat Med 2005; 11: 26–27.
Acknowledgements
JDL is a recipient of the Dana Foundation and Irvington Institute's Human Immunology Fellowship from the Cancer Research Institute and is supported by a Pilot Grant from the National Multiple Sclerosis Society (PP1145) and an Institutional Clinical and Translational Science Pilot and Collaborative Project Grant (to the Rockefeller University Hospital). CM is supported by the Dana Foundation's Neuroimmunology program, the Arnold and Mabel Beckman Foundation, the Alexandrine and Alexander Sinsheimer Foundation, the Burroughs Wellcome Fund, the Starr Foundation, the National Cancer Institute (R01CA108609 and R01CA101741), the National Institute of Allergy and Infectious Diseases (RFP-NIH-NIAID-DAIDS-BAA-06-19), the Foundation for the National Institutes of Health (Grand Challenges in Global Health), and an Institutional Clinical and Translational Science Award (to the Rockefeller University Hospital).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Kroemer
Rights and permissions
About this article
Cite this article
Lünemann, J., Münz, C. Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ 16, 79–86 (2009). https://doi.org/10.1038/cdd.2008.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.113
Keywords
This article is cited by
-
Signaling through NOD-2 and TLR-4 Bolsters the T cell Priming Capability of Dendritic cells by Inducing Autophagy
Scientific Reports (2016)
-
Autophagy: controlling cell fate in rheumatic diseases
Nature Reviews Rheumatology (2016)
-
Infergen Stimulated Macrophages Restrict Mycobacterium tuberculosis Growth by Autophagy and Release of Nitric Oxide
Scientific Reports (2016)
-
Autophagy genes in immunity
Nature Immunology (2009)
-
Autophagy in aging, disease and death: the true identity of a cell death impostor
Cell Death & Differentiation (2009)
