
Abstract
During skeletal muscle growth and regeneration, the majority of differentiating myoblasts undergoes cell–cell fusion to form multinucleated myofibers, whereas a proportion of myoblasts undergoes apoptosis. The treatment of myoblasts with prostaglandin F2α (PGF2α) during myogenesis in vitro leads to the formation of large myotubes, but the mechanism by which PGF2α promotes myotube growth has not been investigated. Here, we demonstrate that PGF2α reduces cell death during myogenesis in vitro and in vivo. In addition, we show that PGF2α increases expression of the inhibitor of apoptosis protein (IAP) BRUCE through a pathway dependent on the nuclear factor of activated T cell 2 transcription factor. Importantly, PGF2α-mediated reduction in muscle cell death is dependent on BRUCE, and overexpression of BRUCE is sufficient to promote muscle cell survival and growth. These results establish a previously unrecognized link between NFAT signaling and regulation of IAP expression and are the first to identify a signaling pathway that increases BRUCE expression. In addition, our results provide evidence that increasing the pool of muscle cells available for fusion by inhibiting cell death enhances myotube growth.
Similar content being viewed by others


Main
During skeletal muscle myogenesis, muscle precursor cells, or myoblasts, undergo a phase of rapid proliferation, followed by cell-cycle exit, myogenic differentiation, and finally cell fusion. These events lead to the formation of multinucleated myofibers in vivo and myotubes in vitro. Skeletal muscle growth and regeneration are dependent on an adequate pool of myoblasts. Therefore, the identification and characterization of molecules that promote the expansion, survival, and fusion of myoblasts are important not only for gaining a better understanding of myogenesis, but also for developing treatments for the loss of muscle mass that occurs with disease and aging.
A number of studies have suggested that prostaglandins (PGs), signaling molecules derived from arachidonic acid by cyclooxygenase (COX), and PG synthase enzymes are important during muscle growth.1 Several PGs have been implicated in multiple phases of myogenesis.1, 2, 3, 4 For example, prostaglandin F2α (PGF2α) is secreted by skeletal muscle cells in vitro and in vivo5, 6 and promotes myotube growth during later stages of myogenesis by increasing the number of nuclei within myotubes.3 PGF2α signals by the FP receptor to increase levels of intracellular calcium, activating the nuclear factor of activated T cells 2 (NFATC2) transcription factor in muscle cells. PGF2α-induced myotube growth is dependent on NFATC2, suggesting that this growth may be dependent on a transcriptional target of NFATC2. A known target of NFATC2 transcription, the cytokine interleukin-4 (IL-4), also promotes muscle growth.7 However, PGF2α does not require IL-4 to exert its growth-promoting effect on muscle cells, as IL-4 null myotubes undergo growth similar to that of wild-type cells in response to PGF2α treatment.8
During myogenesis, a majority of myoblasts exits the cell cycle and undergoes terminal differentiation. Meanwhile, a proportion of differentiating myoblasts undergoes cell death.9, 10, 11, 12, 13 Interestingly, signaling pathways required for the initiation and execution of programmed cell death, or apoptosis, are activated during myogenesis.10, 14, 15, 16 For example, the cysteine protease caspase 3 is not only activated during myogenesis, but also its activity is required for the initiation of myogenic differentiation.14 The mechanism by which the majority of muscle cells undergoes caspase-dependent differentiation but escapes caspase-induced apoptosis is not clear.
Control of apoptosis is critical for development and homeostasis and the balance between life and death is tightly controlled by multiple pathways within the cell. The activity of apoptosis-promoting factors like caspases is in part regulated by a class of proteins known as inhibitor of apoptosis proteins (IAPs).17 Members of the IAP family contain at least one baculovirus IAP repeat (BIR) domain and most also contain domains associated with the ubiquitin degradation pathway. BRUCE (for BIR ubiquitin-conjugating enzyme) is an approximately 530 kDa mouse IAP that is thought to inhibit apoptosis by facilitating the ubiquitin-dependent degradation of multiple caspases.18, 19 BRUCE null mice are embryonic lethal, whereas overexpression of either BRUCE or its human ortholog, Apollon, is sufficient to inhibit apoptosis induced by a variety of stimuli.18, 20, 21, 22 Despite the significant role for BRUCE in blocking apoptosis, very little is known about the regulation of BRUCE expression.
Here, we show that PGF2α reduces cell death during myogenesis and increases expression of BRUCE in an NFATC2-dependent manner. BRUCE is required for PGF2α-mediated muscle cell growth, as siRNA-mediated knockdown of BRUCE expression blocks the ability of PGF2α to induce myotube growth. In addition, overexpression of BRUCE is sufficient to inhibit muscle cell death during myogenesis and thereby leads to the formation of larger myotubes in vitro. These results not only provide important information regarding the mechanism by which PGF2α promotes muscle growth, but also introduce a novel regulator of BRUCE expression.
Results
PGF2α reduces cell death during myogenesis
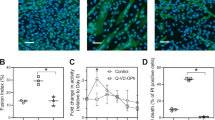
We previously reported that PGF2α promotes myotube growth during myogenesis in vitro.3 To confirm this observation, primary mouse muscle cells were differentiated for 24 h, treated with PGF2α, and assessed for changes in myotube size 24 h after treatment. As expected, cells treated with PGF2α formed larger myotubes after 48 h of differentiation (Figure 1a). In addition, the proportion of myotubes containing ⩾5 nuclei increased with PGF2α treatment (Figure 1b). Control myotubes did not grow as large even at 72 h suggesting that PGF2α does not promote myotube growth by accelerating the rate of growth. Our previous studies indicated PGF2α did not induce gross differences in total DNA content as measured using a spectrophotometer assay.3 However, in the current study we consistently noted small changes in cell density following PGF2α treatment (Figure 1a). Therefore, we hypothesized that the change in myotube size following PGF2α treatment might be a consequence of increasing the number of cells available for fusion. Consistent with this hypothesis, PGF2α treatment led to an 18% increase in the total number of nuclei following 48 h of differentiation (Figure 1c). The change in cell number following PGF2α treatment was not transient, as the total number of nuclei remained elevated, even after 72 h of differentiation (Figure 1c). To determine whether the increase in cell number following PGF2α treatment was a result of a change in the fraction of cells undergoing proliferation, cells were incubated with the thymidine analogue 5-bromo-2-deoxyuridine (BrdU) at the time of PGF2α treatment. After 24 h, cells were assessed for BrdU incorporation. PGF2α did not increase the percentage of BrdU+ cells (Figure 1d), suggesting that PGF2α does not increase myotube size by promoting cell proliferation.

Prostaglandin F2α (PGF2α) reduces cell death during myogenesis. (a) Primary myoblasts were differentiated for 24 h, treated with vehicle (V) or 1 μM PGF2α, and 24 h later fixed and immunostained for eMyHC (bar, 50 μm). (b) Myoblasts were treated as in (a) and fixed after 48 and 72 h of differentiation. The number of nuclei per myotube was counted and the percentage of myotubes containing ⩾5 nuclei was calculated. PGF2α increased the percentage of myotubes with ⩾5 nuclei. Control myotubes did not increase in size with time. (c) The average number of nuclei from five fields of each condition was calculated. After 48 h of differentiation, PGF2α increased the number of nuclei per field by approximately 18%, whereas after 72 h a 22% increase occurred (n=3 at each time point). (d) Primary myoblasts were differentiated for 24 h, treated with V or PGF2α and simultaneously incubated with 5-bromo-2-deoxyuridine (BrdU). Cells were fixed 24 h later, immunostained for BrdU, and nuclei were visualized with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI). No difference was observed in the percentage of BrdU+ nuclei (n=3). (e) Primary myoblasts were differentiated for 24 h, treated with V or PGF2α, and 24 h later dead cells in the media were stained with propidium iodide (PI). PGF2α reduced the number of PI+ cells by 62% (n=3). (f) Myoblasts were differentiated for 24 h at low density, treated with V or PGF2α, and collected 16 h later. Analysis of DNA content by flow cytometry revealed a 25% reduction in sub-diploid DNA content following PGF2α treatment. (g) Schematic representation of myoblast transplantation experiment. Briefly, male myoblasts were pretreated with V or PGF2α 24 h prior to transplant into the tibialis anterior (TA) of recipient female mice. One week after the transplant, DNA from each TA was collected and real-time PCR was performed with primers for a Y-chromosome gene. (h) Pretreatment with PGF2α led to an approximately 2.6-fold increase in male DNA content within the recipient TA muscles 1 week after transplantation (n=11). All data are mean±S.E. (*significantly different, P<0.05)
Because PGF2α increases cell number without altering proliferation, we speculated that PGF2α treatment reduces cell death during myogenesis. To quantify the extent of cell death that occurs during differentiation in vitro, we collected floating cells from the media following 48 h of differentiation and incubated the cells with propidium iodide (PI) to stain dead cells. Approximately 12% of control cells died during this time period (Figure 1e), but PGF2α reduced the number of PI+ cells following 48 h of differentiation by 62% (Figure 1e). To determine whether a proportion of the cell death that occurs during myogenesis results from programmed cell death, or apoptosis, differentiating muscle cells were analyzed for sub-diploid DNA content by flow cytometry. Myoblasts were cultured in differentiation media (DM) at low density for 24 h to induce differentiation but restrict cell fusion, treated with vehicle or PGF2α, and fixed 16 h later. As a positive control for apoptosis, muscle cells were treated with 1 μM staurosporine for 4 h. PI staining was analyzed by flow cytometry and revealed a 25% reduction in apoptosis following PGF2α treatment (Figure 1f). Together, these results indicate that PGF2α reduces apoptosis during myogenesis.
To analyze whether PGF2α could also alter the extent of muscle cell death in vivo, we assessed myoblast survival following transplantation. We chose myoblast transplantation as a model for muscle cell death in vivo because a high percentage of donor myoblasts undergoes death within several days of transplantation into recipient muscle.23 PGF2α-pretreated male myoblasts were transplanted into the tibialis anterior (TA) muscles of female mice (Figure 1g). One week after transplantation, host muscles were collected and the extent of donor cell survival was estimated by performing real-time PCR with a Y chromosome-specific gene. PGF2α pretreatment increased the quantity of male DNA present in host muscles 1 week after transplantation approximately 2.6-fold (Figure 1h), indicating that PGF2α pretreatment increased the number of male donor cells that survived 1 week after transplantation. Although we cannot conclude that PGF2α acts in the same manner in vitro and in vivo, our data do support a model in which PGF2α protects myoblasts from cell death.
PGF2α increases BRUCE expression during myogenesis
To determine the mechanism by which PGF2α mediates myotube growth and muscle cell survival, we performed microarray analyses of RNA isolated from differentiating muscle cells 6 h after treatment with PGF2α. Twenty known genes and several uncharacterized transcripts were identified as differentially expressed in response to PGF2α through this microarray (Figure 2a). We were especially interested in the upregulation of BRUCE mRNA by PGF2α, given the reported role for BRUCE in inhibition of apoptosis.18, 21 Therefore, we confirmed by real-time RT-PCR that BRUCE mRNA expression increases approximately twofold in response to PGF2α treatment (Figure 2b). To determine whether PGF2α treatment increases BRUCE protein expression, we utilized flow cytometry (Figure 2c) and determined that following 6 h of PGF2α treatment, the proportion of BRUCE+ muscle cells increased 9.8±0.9% relative to vehicle (n=4, P<0.001).

Prostaglandin F2α (PGF2α) increases expression of BIR ubiquitin-conjugating enzyme (BRUCE). (a) Primary myoblasts were differentiated for 24 h, treated with vehicle (V) or 1 μM PGF2α, and 6 h later total RNA was collected. Microarray analyses were performed and genes with changes in expression of at least twofold with P<0.05 in response to PGF2α treatment are listed. (b) Real-time RT-PCR confirmed a twofold increase in BRUCE mRNA following 6 h of PGF2α treatment. Data are mean±S.E., n=3. (*significantly different, P<0.05) (c) Representative histograms of BRUCE protein analysis by flow cytometry. The percentage of BRUCE+ cells for each experiment was determined by setting the gate to include <1% of cells incubated with control mouse IgG. PGF2α treatment increased the number of BRUCE+ cells by 9.8±0.9% relative to vehicle (n=4, P<0.001)
Expression of BRUCE during myogenesis has not been previously reported. We therefore performed RT-PCR and western blot analyses to examine BRUCE expression during myogenesis in vitro. BRUCE mRNA was present in proliferating myoblasts (0 h; Figure 3a) and following the onset of differentiation (24 and 48 h). However, BRUCE protein was detected only in differentiating myotubes, and not proliferating myoblasts (Figure 3b), suggesting that post-transcriptional regulation of BRUCE expression may occur in myoblasts. Immunostaining of muscle cells after 48 h of differentiation revealed that BRUCE protein was present in both mononucleated cells and myotubes (Figure 3c). To determine when BRUCE is expressed during myogenesis in vivo, muscle injury was induced by injection of BaCl2 into the TA muscles of adult mice. RNA and protein were collected from uninjured muscles or after 3, 5, 7, or 14 days of regeneration. RT-PCR determined that BRUCE mRNA is expressed throughout myogenesis in vivo (Figure 3d), but BRUCE protein was detected only at later stages of regeneration (Figure 3e). Together, these results demonstrate that BRUCE is expressed during skeletal muscle myogenesis.

BIR ubiquitin-conjugating enzyme (BRUCE) is expressed during myogenesis in vitro and in vivo. (a) Primary myoblasts were differentiated for 0, 24, or 48 h and BRUCE RNA was analyzed by RT-PCR. 18S ribosomal RNA was used as an internal control. BRUCE, 372 bp; 18S, 488 bp. (b) Primary myoblasts were differentiated for 0, 24, or 48 h and BRUCE protein was analyzed by western blot. Ponceau S staining was used as a loading control. BRUCE, 530 kDa. (c) Representative fluorescent and phase-contrast images of muscle cells after 48 h of differentiation that have been immunostained for BRUCE and counterstained with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) (bar, 25 μm). (d) Muscle regeneration was induced by injection of BaCl2 into the tibialis anterior (TA) muscles of adult mice. Total RNA was isolated from uninjured muscles (0) or following 3, 5, 7, or 14 days of regeneration and BRUCE mRNA was analyzed by RT-PCR. cDNA was pooled from 2–3 mice for each time point. 18S ribosomal RNA was used as an internal control. BRUCE, 372 bp; 18S, 488 bp. (e) Protein was isolated from muscle tissue following injury and BRUCE expression was analyzed by western blot. Ponceau S staining was used as a loading control. BRUCE, 530 kDa
NFATC2 is required for upregulation of BRUCE by PGF2α
We have previously shown that PGF2α does not promote growth of NFATC2 null myotubes,3 suggesting that the transcription factor NFATC2 is required for the growth-promoting effects of PGF2α. Although PGF2α treatment of wild-type cells led to an 18% increase in cell number during myogenesis (Figure 1b), no such effect was observed for NFATC2 null cells (Figure 4a). Similarly, PGF2α did not increase expression of BRUCE mRNA in NFATC2 null myotubes (Figure 4b). To determine if basal expression of BRUCE is regulated by NFATC2 in muscle cells, we assessed BRUCE protein levels in wild-type and NFATC2 myotubes after 24 or 48 h of differentiation (Figure 4c). Western blot analyses revealed that expression of BRUCE protein was not significantly altered in the absence of NFATC2. Together, these results indicate that although NFATC2 is not essential for maintenance of basal levels of BRUCE expression during myogenesis, the PGF2α-mediated increase in BRUCE expression and muscle cell number are dependent on NFATC2.

Nuclear factor of activated T cell 2 (NFATC2) is required for the regulation of BIR ubiquitin-conjugating enzyme (BRUCE) expression in muscle cells. (a) NFATC2−/− myoblasts were differentiated for 24 h, treated with vehicle (V) or prostaglandin F2α (PGF2α), and fixed 24 h later. The average number of nuclei from five fields of each condition was calculated. No difference in the number of nuclei per field following PGF2α treatment was observed. (b) NFATC2−/− myoblasts were differentiated for 24 h, treated with V or PGF2α, and 6 h later total RNA was collected. No difference in BRUCE mRNA was detected by real-time RT-PCR. (c) Wild-type or NFATC2−/− myoblasts were differentiated for 24 or 48 h and BRUCE protein was analyzed by western blot. No difference in BRUCE expression was observed between genotypes at either time point. α-Tubulin was used as a loading control. BRUCE, 530 kDa; α-tubulin, 55 kDa. All data are mean±S.E., n=3
BRUCE is required for PGF2-α-mediated reduction in muscle cell death
Because PGF2α increases expression of BRUCE, an inhibitor of apoptosis, we hypothesized that the reduction of muscle cell death induced by PGF2α is dependent on upregulation of BRUCE. To test this hypothesis, we used RNAi to reduce BRUCE expression in primary myoblasts. One of two siRNA oligonucleotides corresponding to unique regions of the BRUCE transcript (BRUCE-1 and BRUCE-2) or a scrambled control siRNA oligonucleotide were transfected into proliferating myoblasts. BRUCE mRNA (Figure 5a) and protein (Figure 5b) were significantly reduced in differentiating cells by both siRNA oligonucleotides. To determine if BRUCE is required for the growth-promoting effects of PGF2α, myoblasts transfected with control or BRUCE siRNA were differentiated for 24 h, treated with vehicle or PGF2α, and fixed 24 h later. Although PGF2α increased the size of control myotubes, cells transfected with either BRUCE siRNA and treated with PGF2α remained similar in size to vehicle-treated cells (Figure 5c). Consistent with this observation, the percentage of BRUCE siRNA-transfected myotubes containing ⩾5 nuclei did not increase in response to PGF2α (Figure 5d). In addition, while PGF2α increased the total number of control cells, BRUCE siRNA blocked this increase in cell number (Figure 5e). PGF2α was also unable to reduce the death of cells transfected with BRUCE siRNA (Figure 5f). Together, these results support a model in which PGF2α increases myotube size and reduces cell death by upregulating expression of BRUCE.

BIR ubiquitin-conjugating enzyme (BRUCE) is required for prostaglandin F2α (PGF2α)-mediated myotube growth. Primary myoblasts were transfected with one of two siRNA oligonucleotides for BRUCE (BRUCE-1 or BRUCE-2) or a scrambled control (C) and switched to differentiation media (DM) 24 h later. (a) After 24 h in DM, BRUCE mRNA was analyzed by real-time RT-PCR. BRUCE mRNA expression was reduced greater than 70% by both siRNAs. (b) Western blots were performed with protein from transfected cells after 24 h in DM, which revealed that BRUCE protein expression was reduced by both siRNAs. α-Tubulin was used as a loading control. BRUCE, 530 kDa; α-tubulin, 55 kDa. (c) Myoblasts were differentiated for 24 h, treated with V or 1 μM PGF2α, and 24 h later fixed and immunostained for eMyHC (bar, 50 μm). (d) The number of nuclei per myotube was counted and the percentage of myotubes containing ⩾5 nuclei was calculated. Although PGF2α significantly increased the percentage of myotubes containing ⩾5 nuclei, BRUCE siRNA blocked this increase in myotube size. (e) The average number of nuclei from five fields of each condition was calculated. Results are presented as the percentage difference between PGF2α- and V-treated cells. Although PGF2α increased the number of control nuclei by approximately 20%, no such increase was observed for BRUCE siRNA-transfected cells. (f) Dead cells in the media were collected and stained with propidium iodide (PI) after 48 h in DM. Although the number of PI+ control-transfected cells was reduced by 45% following PGF2α treatment, PGF2α did not alter the death of BRUCE siRNA-transfected cells. All data are mean±S.E., n=3. (*significantly different, P<0.05)
Overexpression of BRUCE in muscle cells is sufficient to promote myotube growth and reduce cell death during myogenesis
If increased expression of BRUCE is required for PGF2α to promote myotube growth, we speculated that increasing expression of BRUCE in the absence of PGF2α would be sufficient to reduce muscle cell death and increase myotube size. To test this hypothesis, primary myoblasts transfected with either a BRUCE expression vector18 or a control vector were differentiated for 48 h. Western blot analysis of protein lysates collected after 24 h of differentiation indicated that BRUCE was highly expressed in cells that had been transfected with the BRUCE expression vector (Figure 6a). BRUCE-overexpressing myotubes were larger (Figure 6b) and the proportion of myotubes containing ⩾5 nuclei increased 28% over control (Figure 6c). In addition, overexpression of BRUCE increased the total number of nuclei by 22% (Figure 6d) and reduced cell death after 48 h of differentiation by 36% (Figure 6e). To determine if increasing BRUCE expression reduces cell death in vivo, we transplanted male muscle cells overexpressing BRUCE into the TA muscles of female mice. At 1 week after transplantation, the quantity of male DNA present in the host muscles that received BRUCE-overexpressing myoblasts was approximately eightfold higher than muscles that received control myoblasts (Figure 6f), suggesting that BRUCE expression increased muscle cell survival in vivo. Together, these results demonstrate that increasing expression of BRUCE is sufficient to promote myotube growth and reduce cell death during myogenesis.

BIR ubiquitin-conjugating enzyme (BRUCE) overexpression is sufficient to promote myotube growth and reduce muscle cell death during myogenesis. (a) Primary myoblasts transfected with control (C) or BRUCE expression vectors were differentiated for 24 h and BRUCE protein was analyzed by western blot. BRUCE, 530 kDa. (b) Primary myoblasts transfected with C or BRUCE expression vectors were differentiated for 48 h, fixed, and immunostained for eMyHC (bar, 50 μm). (c) The number of nuclei per myotube was counted and the percentage of myotubes containing ⩾5 nuclei was calculated. Overexpression of BRUCE increased the proportion of myotubes with ⩾5 nuclei 28% over control. (d) The average number of nuclei from five fields of each condition was calculated. BRUCE overexpression increased the number of nuclei by 22%. (e) The number of propidium iodide (PI)+ cells in the media of BRUCE-overexpressing myotubes was reduced 36% compared with control. (f) Male myoblasts were transfected with a control or BRUCE expression vector and transplanted into the tibialis anterior (TA) of recipient female mice. Real-time PCR of a Y-chromosome gene demonstrated that male DNA content within the host muscle 1 week after transplant was increased approximately eightfold by BRUCE overexpression in donor muscle cells. All data are mean S.E., n=3. (*significantly different, P<0.05)
Discussion
During muscle development and regeneration from injury in vivo, a subpopulation of myoblasts undergoes apoptosis. 11, 12, 24, 25 During myogenesis in vitro, as many as 50% of myoblasts reportedly undergo apoptosis,9, 10, 15 although the extent of cell death likely depends on origin of the myoblasts and the culture conditions used for each study. Myoblasts may undergo apoptosis during myogenesis as a means of regulating muscle size. Accordingly, excess myoblasts may be eliminated by apoptosis when the muscle reaches a predetermined size. Alternatively, a portion of myoblasts may undergo apoptosis as a consequence of the cellular stress involved in transitioning from a highly proliferative to terminally differentiated cell type. Our data show that PGF2α reduces the extent of apoptosis that occurs during myogenesis in vitro (Figure 1f). Furthermore, our transplantation results suggest that PGF2α treatment promotes muscle cell survival in vivo (Figure 1h). PGF2α also increases myotube size (Figure 1a;3), suggesting that reducing cell death during myogenesis increases the pool of myoblasts available for fusion, and thus enhances myotube growth. In support of this concept, overexpression of Necdin reduces the number of apoptotic cells during skeletal muscle regeneration and promotes the formation of larger myofibers.24 Therefore, a change in the proportion of myoblasts that undergoes apoptosis during myogenesis influences muscle size.
PGs have been implicated in myoblast proliferation, differentiation, migration, and fusion,1 but the current study is the first study to demonstrate a function for a PG in myoblast survival. Studies in other cell types, however, have linked PG signaling to a reduction in apoptosis. Treatment of several cell types with PGF2α reduces cell death in response to apoptotic stimuli,26, 27, 28 but induces apoptosis in others.29 Thus, the relationship between PG signaling and apoptosis is cell type specific. Interestingly, the deregulation of apoptosis that occurs with tumorogenesis is often accompanied by increased expression of COX-2 and PGs.30 Our results provide an additional link between PG signaling and the inhibition of cell death and may have important implications for therapeutic strategies to control the reduction of apoptosis associated with tumorogenesis. COX-2 inhibitors have been the subjects of clinical trials for the prevention of cancer, but the increased rates of cardiovascular complications associated with the use of COX-2 inhibitors have raised serious concerns. Selective inhibitors of specific PGs may reduce the broad range of effects caused by COX-2 inhibition and therefore may be better candidates for chemoprevention.
Control of apoptosis is critical for tissue development and maintenance. The two convergent signaling cascades, the extrinsic and intrinsic pathways, which initiate apoptosis have been studied extensively. Both pathways lead to the activation of the caspase enzymes by cleavage of pro-caspase zymogens. The protease activity of caspases ultimately results in the execution of cell death. Interestingly, the activation of several caspases occurs during myogenesis.14, 15, 31 Caspase-3 is activated early in muscle differentiation and promotes the activation of upstream regulators of the MAP kinase p38, a key initiator of myogenic differentiation.14 Caspase-12 is also activated during muscle differentiation, and has been implicated in the initiation of apoptosis that occurs during myogenesis.15 Activated caspases are inhibited by the IAP family of proteins, thus the IAPs serve as a shield to the final execution of apoptosis.17 Expression levels of IAPs are dynamic, and can be regulated at the transcriptional, post-transcriptional, and post-translational levels.17 Interestingly, increased expression of IAPs has been reported in several types of cancer cells and tumors.17 Although regulation of IAP transcription has not been well characterized, expression of the IAP survivin is cell cycle dependent32 and NF-κB can modulate expression of c-IAP2 and xIAP.33, 34 Interestingly, PGE2 increases expression of survivin in multiple cell types, likely by activation of the E prostanoid (EP)2/EP4 receptor and downstream of phosphatidylinositol-3-kinase activity.35, 36 Here, we have shown that PGF2α increases expression of another IAP, BRUCE (Figure 2).
BRUCE, also known as Apollon in humans, is a 530 kDa mouse protein that is expressed in a variety of tissues.19 BRUCE not only binds caspases, but also promotes degradation of caspases by the ubiquitin proteasome pathway.18, 20 BRUCE/Apollon expression protects cells from a number of apoptotic stimuli.18, 20 Interestingly, caspase activity during spermatogenesis is regulated by the Drosophila homologue of BRUCE, which protects the differentiating sperm from apoptosis.37 Multiple groups have generated BRUCE-deficient mice, and all mice are embryonic lethal.20, 21, 22 However, whether embryonic lethality associated with loss of BRUCE function is caused by an increase in apoptosis is unclear. Ren et al.22 reported the abundance of apoptotic cells in the placenta and yolk sac in mice lacking the C-terminal half of BRUCE. Surprisingly however, Lotz et al. did not detect significant changes in the extent of apoptosis in the developing BRUCE null embryo. Embryonic lethality in the BRUCE null mice may arise from a potential role for BRUCE in the regulation of cell proliferation.21, 38 In our hands, siRNA knockdown of BRUCE in myoblasts does not alter cell proliferation (data not shown). However, siRNA knockdown of BRUCE expression blocks the ability of PGF2α to reduce cell death during myogenesis (Figure 5). Unexpectedly, knockdown of BRUCE does not significantly alter basal levels of cell death. Possibly, other IAPs may compensate for this decrease in BRUCE expression during embryogenesis and myogenesis, thereby protecting cells from death in the absence of BRUCE function. Importantly, overexpression of BRUCE in myoblasts is sufficient to protect cells from undergoing death during myogenesis and leads to the formation of larger myotubes (Figure 6). Our results are consistent with previous studies and suggest that although loss of BRUCE does not induce spontaneous apoptosis, increasing BRUCE expression above basal levels is sufficient to protect cells from a variety of apoptotic stimuli.
Despite the importance of BRUCE in the inhibition of apoptosis, signaling pathways involved in the regulation of BRUCE expression have not been identified. Our results suggest that NFATC2, a transcription factor that is activated in response to increases in intracellular Ca2+ levels, plays a key role in the regulation of BRUCE expression in muscle cells. PGF2α signals through the G-protein-coupled FP receptor to activate intracellular Ca2+ signaling.35 Previously, we showed that treating myotubes with PGF2α stimulates NFATC2 translocation to the nucleus.3 Here, we have demonstrated that PGF2α increases cell number and BRUCE expression, but only in the presence of NFATC2 (Figure 4). NFATC2 is not required for basal BRUCE expression during myogenesis. Interestingly, this is not the first example of gene expression that is regulated in an induced, but not basal manner by NFATC2. For example, although NFATC2 is required for increased expression of CD95 ligand in response to T-cell receptor activation in T cells, constitutive expression of CD95 ligand in Sertoli cells is not dependent on NFATC2.39 Two NFAT-binding sites in the CD95 ligand promoter direct inducible, but not constitutive gene expression. Whether the transcriptional activity of NFATC2 directly regulates BRUCE expression in muscle, or if an intermediate pathway is involved, remains to be determined once the promoter region of BRUCE is defined. Importantly, this work identifies a hitherto unrecognized relationship between NFAT signaling and regulation of IAP expression.
The identification of factors that increase the pool of myoblasts available for fusion and therefore promote muscle growth may lead to novel therapies for the treatment of muscle disorders. We have shown that PGF2α and BRUCE support myoblast survival in vitro and in vivo, which may be valuable for increasing the success of myoblast transplantation strategies for muscular disorders. In addition, this work has identified PGF2α and NFATC2 as novel regulators of BRUCE expression. To date, few factors regulating IAP expression have been identified and the discovery of additional regulators could provide new targets for the treatment of cancer.
Materials and Methods
Primary muscle cell culture
Primary myoblasts were derived from the hindlimb muscles of 6- to 8-week-old female wild-type or NFATC2−/− Balb/C mice, unless otherwise noted, and cultured to >99% purity as previously described.7 Cells were maintained in growth media (GM: Ham's F10, 20% FBS, 5 ng/ml bFGF, 100 U/ml penicillin G, 100 μg/ml streptomycin) in a humidified 5% CO2 incubator at 37°C on collagen-coated dishes. Cells were differentiated in DM (DMEM, 1% Insulin-Transferrin-Selenium A (Invitrogen, Carlsbad, CA, USA) 100 U/ml penicillin G, 100 μg/ml streptomycin) on six-well plates coated with Entactin-Collagen IV-Laminin (E-C-L; Millipore, Billerica, MA, USA) at a density of 2 × 105 cells per well. For PG experiments, myoblasts were differentiated for 24 h prior to treatment with vehicle (0.095% ethanol) or 1 μM PGF2α (Sigma, St. Louis, MO, USA).
Analysis of cell proliferation, death, and fusion
To assess cell proliferation, differentiating myotubes were incubated in 25 μM BrdU (Sigma) for 24 h. Cells were subsequently fixed in 2% paraformaldehyde and immunostained using a BrdU antibody (Accurate Chemical and Scientific Corp., Westbury, NY, USA) at a dilution of 1 : 500. At least 500 myoblasts from three independent isolates were analyzed for each condition.
To analyze cell death, the medium was collected following 48 h of differentiation and spun at 1400 g for 2 min to collect floating cells. These cells were then resuspended in PBS containing 0.5 μg/ml PI (Sigma) and incubated for 5 min. PI+ cells were counted on a hemocytometer to quantify the number of dead cells in each condition. Apoptosis of differentiating muscle cells was analyzed as described previously.40, 41 Myoblasts were differentiated at low density (106 cells per 150 mm dish) for 40 h. The medium was collected and spun at 1400 g for 2 min to collect floating cells and adherent cells were collected by trypsinization. Cells were fixed for 1 h in 100% ethanol on ice and subsequently suspended in 50 mM Na2HPO4 and 10 mM citric acid, pH 7.8, diluted in PBS for 5 min at room temperature. Cells were then suspended in 0.5 μg/ml PI and analyzed by flow cytometry. Data from all flow cytometry experiments were analyzed using FlowJo software (Version 8.7, TreeStar Inc., Ashland, OR).
To assess myoblast fusion, cells were differentiated for 48 h, fixed in 3.7% formaldehyde, and immunostained for embryonic myosin heavy chain (F1.652, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) as described previously.3 The number of nuclei in each myotube (⩾2 nuclei) was counted and the percentage of myotubes with ⩾5 nuclei was calculated. Fusion assays were performed using myoblasts from three independent isolates and ⩾100 myotubes were analyzed for each experiment.
Myoblast transplantations
Primary wild-type myoblasts from 6- to 8-week-old male C57Bl/6 mice were prepared for transplantation by differentiating at low density (106 cells per 150 mm E-C-L-coated plate) for 24 h while being treated with vehicle or 1 μM PGF2α at 0 and 12 h of differentiation. Alternatively, myoblasts transfected with control or BRUCE vectors (as described below) were differentiated at low density for 24 h. Prior to transplantation, the cells were suspended at a concentration of 6x104 cells/μl in PBS with 0.1% bovine serum albumin (BSA). A 25 μl Hamilton syringe was used to inject a 5 μl cell suspension into the TA muscles of 8-week-old female C57BL/10ScSn-Dmdmdx/J mice (Jackson Labs, Bar Harbor, ME, USA). At 7 days after transplantation, TA muscles were collected and DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA isolated from each muscle was suspended in 200 μl elution buffer.
Real-time PCR was used to quantify male DNA content in each recipient female muscle using the iCycler iQ Real-Time Detection system (Bio-Rad, Hercules, CA, USA). PCR reactions were performed using 4 μl DNA with 6 min denaturation at 95°C followed by 45 cycles of 95°C for 15 s, 58°C for 20 s, and 60°C for 35 s. SYBR Green fluorescence was measured after the end of the extension step of each cycle. Primers for the Y chromosome-specific gene Zfy1 were used to amplify the male DNA (accession no. NM_009570; sense, 5′-TGGAGAGCCACAAGCTAACCA; antisense, 5′-TCCCAGCATGAGAAAGATTCTTC). The starting quantity of male DNA template in each sample was determined by comparing the threshold cycle of PCR amplification (CT) to a standard curve. To normalize samples for total DNA content, real-time PCR was performed with primers for an autosomal gene (NFAT5, accession no. NM_018823; sense, 5′-GGGCTATAGACATGCACCACCACACAG; antisense, 5′-AAGGGCTTCTTCCAGAATGG). All real-time PCR reactions were performed in triplicate.
Microarray analysis
Primary myoblasts were differentiated for 24 h prior to treatment with vehicle (0.095% ethanol) or 1 μM PGF2α (Sigma). At 6 h after treatment, RNA was isolated in a two-step process. Total RNA was first isolated using TRIzol reagent (Invitrogen), followed by further purification using the RNeasy kit and DNase treatment (Qiagen) according to the manufacturer's instructions. RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA integrity number of all samples was above 9.0. The Chemiluminescent RT-IVT Labeling Kit (Applied Biosystems, Foster City, CA, USA) was used to amplify 1 μg of total RNA according to the manufacturer's instructions. For each sample, 10 μg of cRNA was hybridized to a Mouse Genome Survey Microarray (Applied Biosystems) and the 1700 Chemiluminescent Microarray Analyzer (Applied Biosystems) was used to scan the microarrays and generate raw data. Quantile normalization of all data was performed using Bioconductor (www.bioconductor.org) and probes with the signal to noise ratio higher than 3 and flags lower than 5000 were used for further analysis. Probes with P<0.05 and a fold change of ⩾2 were considered significant. RNA was isolated from three independent cell isolates. Microarray analyses were performed by the Winship Cancer Institute Microarray Core at Emory University.
Analysis of BRUCE mRNA expression
Muscle regeneration was induced in the TA muscles of 8-week-old female Balb/C mice by injection of 50 μl of 1.2% BaCl2. Differentiation of primary muscle cells was performed as described above. Total RNA was isolated from tissue or cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions and cDNA was generated by reverse transcription using 1.5 μg of total RNA. For semiquantitative RT-PCR, BRUCE cDNA was amplified by 5 min denaturation at 95°C, followed by 32 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s. Primers specific to BRUCE (accession no. NM_007566; sense, 5′-CTCAGTCAGTCCTGCCTCATC; antisense, 5′-CTCAGCAGTCCTCTGGATGTC) were designed to span intron and exon boundaries to control for any contaminating DNA in the RNA samples. Amplicons were separated by electrophoresis in a 1% agarose gel and visualized with ethidium bromide. To analyze BRUCE mRNA expression by quantitative real-time RT-PCR, BRUCE cDNA standards were first generated by performing RT-PCR with primers described above and gel purified with the QIAquick gel extraction kit (Qiagen). Real-time PCR was performed as described above with 40 cycles of 95°C for 30 s, 65°C for 30 s, and 72°C for 30 s. The primers used for real-time RT-PCR amplified a region of the BRUCE transcript within the amplicon that had been generated for the standard curve (sense, 5′-ATCGTGCTCTGCTAGAGTTGC; antisense, 5′-ATCTTAGGCGGTTGGTGTAGG). Amplification of 18S ribosomal RNA using QuantumRNA primers (Ambion, Foster City, CA, USA) was used an input control for all reactions. Total RNA was isolated from three independent cell isolates and all real-time PCR reactions were run in triplicate.
Analysis of BRUCE protein expression
For flow cytometry analysis, myoblasts were differentiated at low density (106 cells per 150 mm dish) for 24 h, treated with vehicle or PGF2α, and collected by trypsinization 6 h later. Cells were fixed in 100% methanol for 10 min on ice and subsequently washed with PBS containing 0.1% BSA. Cells were then incubated for 30 min on ice in BRUCE antibody (BD, Franklin Lakes, NJ, USA) or control mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA) diluted to a concentration of 2.5 μg/ml. Following three washes in PBS, cells were incubated on ice for 30 min in R-phycoerythrin-labeled goat anti-mouse (Invitrogen) antibody diluted at a ratio of 1 : 250. Following three additional washes, cells were analyzed by flow cytometry.
For western blot analysis, total protein was isolated from primary muscle cells or muscle tissue using M-PER reagent (Pierce, Rockford, IL, USA) supplemented with protease inhibitors (Mini complete; Roche, Basel, Switzerland). Cells were lysed at 4°C for 15 min and lysates were cleared of insoluble material by spinning at 21 000 g for 15 min. Protein concentration was determined by Bradford analysis (Bio-Rad) and equal amounts of protein (⩾15 μg) were loaded on each lane of a 5% polyacrylamide gel. Samples were separated by SDS-PAGE and transferred at 400 mA for 2 h to a nitrocellulose membrane. Immunoblotting was performed using BRUCE (BD) or α-tubulin (Sigma) antibodies diluted at a ratio of 1 : 500.
For immunostaining, primary muscle cells were differentiated for 0, 24, or 48 h and fixed in 3.7% formaldehyde for 10 min. Cells were first incubated for 1 h in block buffer (5% donkey serum in PBS with 0.25% Triton X-100), followed by BRUCE or control IgG antibody diluted at a ratio of 1 : 100 in block buffer for 1 h at room temperature. Following three washes in PBS+0.2% Tween 20 (PBS-T), biotin-conjugated goat anti-mouse IgG antibody (Jackson ImmunoResearch) diluted at a ratio of 1 : 250 in block buffer was added to cells for 1 h and subsequently washed as before. Antibody binding was detected with Texas Red-streptavidin (Jackson ImmunoResearch) diluted at a ratio of 1 : 500 in block buffer. Nuclei were visualized with 4,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma).
Transfection of primary myoblasts
Stealth RNAi (Invitrogen) was used for siRNA ‘knockdown’ experiments. Myoblasts were plated in GM at a density of 105 cells per well on a collagen-coated six-well plate and after 24 h, duplexed RNA was added at a dilution of 80 nM. Cells were then transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 24 h after the transfection, cells were replated and differentiated as described above. Total RNA and protein were isolated after 24 h in DM and knockdown was assessed by real-time PCR and western blot. Two siRNA oligonucleotides corresponding to base-pairs 9676–9700 (BRUCE-1, (5′–3′) CCUGACAAUGCAGAAGGAAUCCAUA) or 11313–11337 (BRUCE-2, (5′–3′) GAAAGAUUGGCUUGGUGGUUCUGAA) of the BRUCE mRNA transcript or a control scrambled oligonucleotide (Stealth RNAi Low GC negative control) were used for each transfection. Knockdowns were performed with myoblasts from three independent cell isolates.
A mammalian expression vector encoding FLAG-tagged full-length BRUCE cDNA18 was used to overexpress BRUCE in primary myoblasts. To select for transfected myoblasts, the cells were co-transfected with a plasmid containing a neomycin resistance gene (pEXPR-IBA3; IBA Biotagnology, Hanover, Germany) at a ratio of nine parts BRUCE-FLAG to one part pEXPR. Lipofectamine LTX with Plus Reagent (Invitrogen) was used to transfect 2 × 105 cells with 2.5 μg total DNA according to the manufacturer's instructions. Cells were subsequently grown in GM containing 50 μg/ml G418 (Invitrogen) for approximately 2 weeks to ensure adequate selection and to allow for recovery from transfection. Overexpression was confirmed by western blot using BRUCE or FLAG (Sigma) antibodies diluted at a ratio of 1 : 500. Transfections were performed with myoblasts from three independent cell isolates.
Statistical analyses
Comparisons between two groups were made using t-tests. Analyses of multiple groups were performed using a two-way ANOVA with Bonferroni's post-test. Statistical analyses were performed using GraphPad Prism 4.0 (GraphPad) for Macintosh or Sigma-Stat 2.03 (SPSS). For all statistical tests, a confidence level of P<0.05 was accepted for statistical significance.
Abbreviations
- PGF2α:
-
prostaglandin F2α
- IAP:
-
inhibitor of apoptosis protein
- NFAT:
-
nuclear factor of activated T cells
- PG:
-
prostaglandin
- COX:
-
cyclooxygenase
- IL-4:
-
interleukin-4
- BIR:
-
baculovirus IAP repeat
- BrdU:
-
5-bromo-2-deoxyuridine
- TA:
-
tibialis anterior
- PI:
-
propidium iodide
References
Prisk V, Huard J . Muscle injuries and repair: the role of prostaglandins and inflammation. Histol Histopathol 2003; 18: 1243–1256.
Shen W, Prisk V, Li Y, Foster W, Huard J . Inhibited skeletal muscle healing in cyclooxygenase-2 gene-deficient mice: the role of PGE2 and PGF2alpha. J Appl Physiol 2006; 101: 1215–1221.
Bondesen BA, Jones KA, Glasgow WC, Pavlath GK . Inhibition of myoblast migration by prostacyclin is associated with enhanced cell fusion. FASEB J 2007; 21: 3338–3345.
Horsley V, Pavlath GK . Prostaglandin F2(alpha) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J Cell Biol 2003; 161: 111–118.
Nowak J, Bohman SO, Alster P, Berlin T, Cronestrand R, Sonnenfeld T . Biosynthesis of prostaglandins in microsomes of human skeletal muscle and kidney. Prostaglandins, LeukotMed 1983; 11: 269–279.
Rossi MJ, Clark MA, Steiner SM . Possible role of prostaglandins in the regulation of mouse myoblasts. J Cell Physiol 1989; 141: 142–147.
Horsley V, Jansen KM, Mills ST, Pavlath GK . IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 2003; 113: 483–494.
Pavlath GK, Horsley V . Cell fusion in skeletal muscle – central role of NFATC2 in regulating muscle cell size. Cell Cycle (Georgetown, Tex) 2003; 2: 420–423.
Chinni C, de Niese MR, Tew DJ, Jenkins AL, Bottomley SP, Mackie EJ . Thrombin, a survival factor for cultured myoblasts. J Biol Chem 1999; 274: 9169–9174.
Dee K, Freer M, Mei Y, Weyman CM . Apoptosis coincident with the differentiation of skeletal myoblasts is delayed by caspase 3 inhibition and abrogated by MEK-independent constitutive Ras signaling. Cell Death Differ 2002; 9: 209–218.
Fidzianska A, Goebel HH . Human ontogenesis. 3. Cell death in fetal muscle. Acta Neuropathologica 1991; 81: 572–577.
McClearn D, Medville R, Noden D . Muscle cell death during the development of head and neck muscles in the chick embryo. Dev Dyn 1995; 202: 365–377.
Wang J, Walsh K . Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science (New York, NY) 1996; 273: 359–361.
Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA . Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA 2002; 99: 11025–11030.
Nakanishi K, Sudo T, Morishima N . Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 2005; 169: 555–560.
O’Flaherty J, Mei Y, Freer M, Weyman CM . Signaling through the TRAIL receptor DR5/FADD pathway plays a role in the apoptosis associated with skeletal myoblast differentiation. Apoptosis 2006; 11: 2103–2113.
Salvesen GS, Duckett CS . IAP proteins: blocking the road to death's door. Nat Rev 2002; 3: 401–410.
Bartke T, Pohl C, Pyrowolakis G, Jentsch S . Dual role of BRUCE as an antiapoptotic IAP and a chimeric E2/E3 ubiquitin ligase. Mol Cell 2004; 14: 801–811.
Hauser HP, Bardroff M, Pyrowolakis G, Jentsch S . A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J Cell Biol 1998; 141: 1415–1422.
Hao Y, Sekine K, Kawabata A, Nakamura H, Ishioka T, Ohata H et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol 2004; 6: 849–860.
Lotz K, Pyrowolakis G, Jentsch S . BRUCE, a giant E2/E3 ubiquitin ligase and inhibitor of apoptosis protein of the trans-Golgi network, is required for normal placenta development and mouse survival. Mol Cell Biol 2004; 24: 9339–9350.
Ren J, Shi M, Liu R, Yang QH, Johnson T, Skarnes WC et al. The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc Natl Acad Sci USA 2005; 102: 565–570.
Skuk D, Caron NJ, Goulet M, Roy B, Tremblay JP . Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol 2003; 62: 951–967.
Deponti D, Francois S, Baesso S, Sciorati C, Innocenzi A, Broccoli V et al. Necdin mediates skeletal muscle regeneration by promoting myoblast survival and differentiation. J Cell Biol 2007; 179: 305–319.
Thomazy VA, Davies PJ . Expression of tissue transglutaminase in the developing chicken limb is associated both with apoptosis and endochondral ossification. Cell Death Differ 1999; 6: 146–154.
Kawamura T, Horie S, Maruyama T, Akira T, Imagawa T, Nakamura N . Prostaglandin E1 transported into cells blocks the apoptotic signals induced by nerve growth factor deprivation. J Neurochem 1999; 72: 1907–1914.
Kroll B, Kunz S, Tu N, Schwarz LR . Inhibition of transforming growth factor-beta1 and UV light-induced apoptosis by prostanoids in primary cultures of rat hepatocytes. Toxicol Appl Pharmacol 1998; 152: 240–250.
Ottonello L, Gonella R, Dapino P, Sacchetti C, Dallegri F . Prostaglandin E2 inhibits apoptosis in human neutrophilic polymorphonuclear leukocytes: role of intracellular cyclic AMP levels. Exp Hematol 1998; 26: 895–902.
Juengel JL, Garverick HA, Johnson AL, Youngquist RS, Smith MF . Apoptosis during luteal regression in cattle. Endocrinology 1993; 132: 249–254.
Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN . Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol 2005; 23: 254–266.
Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y . An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 2002; 277: 34287–34294.
Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998; 396: 580–584.
Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW . Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA 1997; 94: 10057–10062.
Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J . Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med 1998; 188: 211–216.
Krysan K, Merchant FH, Zhu L, Dohadwala M, Luo J, Lin Y et al. COX-2-dependent stabilization of survivin in non-small cell lung cancer. FASEB J 2004; 18: 206–208.
Baratelli F, Krysan K, Heuze-Vourc’h N, Zhu L, Escuadro B, Sharma S et al. PGE2 confers survivin-dependent apoptosis resistance in human monocyte-derived dendritic cells. J Leukoc Biol 2005; 78: 555–564.
Arama E, Agapite J, Steller H . Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev Cell 2003; 4: 687–697.
Pohl C, Jentsch S . Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell 2008; 132: 832–845.
Latinis KM, Norian LA, Eliason SL, Koretzky GA . Two NFAT transcription factor binding sites participate in the regulation of CD95 (Fas) ligand expression in activated human T cells. J Biol Chem 1997; 272: 31427–31434.
Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P et al. Features of apoptotic cells measured by flow cytometry. Cytometry 1992; 13: 795–808.
Vitale M, Zamai L, Mazzotti G, Cataldi A, Falcieri E . Differential kinetics of propidium iodide uptake in apoptotic and necrotic thymocytes. Histochemistry 1993; 100: 223–229.
Acknowledgements
We thank Dr. Maja Ordanic Kodani and the Winship Cancer Institute Microarray Core at Emory University for performing microarray analyses, Yusra Ahmad for assistance with immunocytochemistry, Dr. Navaline Quach for advice on siRNA knockdowns, and Dr. Stefan Jentsch for the BRUCE construct. GKP is supported by grants from National Institutes of Health (AR-052730, AR-047314, and AR-051372) and Muscular Dystrophy Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by S Kumar
Rights and permissions
About this article
Cite this article
Jansen, K., Pavlath, G. Prostaglandin F2α promotes muscle cell survival and growth through upregulation of the inhibitor of apoptosis protein BRUCE. Cell Death Differ 15, 1619–1628 (2008). https://doi.org/10.1038/cdd.2008.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.90
Keywords
This article is cited by
-
Mitochondrial impairment but not peripheral inflammation predicts greater Gulf War illness severity
Scientific Reports (2023)
-
Statin-induced myopathic changes in primary human muscle cells and reversal by a prostaglandin F2 alpha analogue
Scientific Reports (2020)
-
Dual Oxidase Maturation factor 1 (DUOXA1) overexpression increases reactive oxygen species production and inhibits murine muscle satellite cell differentiation
Cell Communication and Signaling (2014)
-
The skeletal muscle arachidonic acid cascade in health and inflammatory disease
Nature Reviews Rheumatology (2014)
-
Inhibition of caspase mediated apoptosis restores muscle function after crush injury in rat skeletal muscle
Apoptosis (2012)
