
Abstract
Mitochondrial Ca2+ homeostasis is today at the center of wide interest in the scientific community because of its role both in the modulation of numerous physiological responses and because of its involvement in cell death. In this review, we briefly summarize a few basic features of mitochondrial Ca2+ handling in vitro and within living cells, and its involvement in the modulation of Ca2+-dependent signaling. We then discuss the role of mitochondrial Ca2+ in the control of apoptotic death, focusing in particular on the effects of pro- and anti-apoptotic proteins of the Bcl-2 family. Finally, the potential involvement of Ca2+ and mitochondria in the development of two diseases, Ullrich muscular dystrophy and familial Alzheimer's disease, is briefly discussed.
Similar content being viewed by others


General Features of Ca2+ Handling by Mitochondria
The ability of isolated mitochondria to take up Ca2+ from the medium and to accumulate it in their matrix in an energy dependent way was first described over 40 years ago.1 At that time, the chemiosmotic hypothesis was still not widely accepted by the scientific community. It was thought that the driving force for Ca2+ accumulation was provided by an unknown high-energy phosphorylated intermediate, X∼P, generated by the electron flow through the respiratory chain or by the hydrolysis of ATP. Later, in the 1970s, the idea that the energy derived from substrate oxidation or by ATP hydrolysis is transformed in a H+ gradient across the inner membrane became widely accepted and was honored by a Nobel Prize to Peter Mitchell in 1978. The concept that Ca2+ uptake by energized mitochondria is driven by the membrane potential across the inner membrane (negative inside) was thus clarified. Acceptance of this idea led to the prediction that, at equilibrium, the accumulation ratio between matrix and extracellular [Ca2+] should only be dictated by the Nernst equation. An inevitable corollary was that with a membrane potential of 180 mV, a divalent cation should accumulate within the matrix with a concentration 1 million fold that of the cytosol, that is for a cytosolic [Ca2+] ([Ca2+]c) of 100 nM, a matrix [Ca2+] ([Ca2+]m) of 0.1 M would be expected. This was clearly contrary to all experimental data and unquestionably incompatible with cell physiology. The paradox was eventually solved when it was demonstrated that the accumulation of Ca2+ within the mitochondrial matrix depends not only on the existence of an electrogenic uniporter (that tends to bring Ca2+ to an electrochemical equilibrium), but also on antiporters (Na+ or H+/Ca2+), driven by the ion gradients, that extrude Ca2+ from the matrix.2 A steady-state [Ca2+]m is reached, far from electrochemical equilibrium, when the rate of Ca2+ influx through the uniporter equals the rate of Ca2+ efflux through the antiporters. The futile Ca2+ cycle occurring continuously across the mitochondrial inner membrane represents a small energy drain, but it is reduced to a minimum under physiological conditions because the ‘affinity’ for Ca2+ of the antiporter is low. In particular, the dependence of the uptake rate on the [Ca2+], at physiological Mg2+ concentration, is highly cooperative and the apparent KM under these conditions is around 20 μM Ca2+. Accordingly, the rate of Ca2+ influx at resting cytosolic [Ca2+] is ∼0.1% of the maximal uptake rate. The functional consequences of this kinetic model are valid to date; any change in the speed of the influx or efflux pathways must lead to changes in the [Ca2+] within the matrix. It is often erroneously assumed that microdomains of high Ca2+ in the cytosol are necessary for driving Ca2+ accumulation by mitochondria in living cells (see below). This is clearly a misinterpretation of the microdomain hypothesis that was formulated to explain the fast Ca2+ uptake by mitochondria in living cells. In fact, no matter how small the rise of [Ca2+]c, an increase should occur also in the mitochondrial matrix (for a review, see Rizzuto and Pozzan3).
Regarding the factors that modulate Ca2+ influx (Vmax or KM) through the uniporter, by far the most important is the extramitochondrial [Ca2+]. Changes in the [Ca2+]c in a living cell occur both under physiological or pathological conditions upon opening of plasma membrane or intracellular Ca2+ channels. Polyamines, such as spermine or cadaverine, slightly increase the KM, but this is observed only at Ca2+ concentrations well above the physiological ones. Recently (see also below), it has been suggested that the uniporter rate (or better, its affinity for Ca2+) can be modulated (increased or decreased) by protein kinases, in particular by p38 MAP kinases (inactivation),4 or protein kinase C (the ζ isoform activates, whereas the β/δ isoforms inactivate it5), although this modulation still requires further clarification. A highly interesting, yet largely neglected phenomenon, is the Ca2+-induced activation of Ca2+ uptake, a phenomenon initially described 20 years ago by Kroner6 and recently re-visited by Moreau et al.7 It consists in a substantial acceleration of the Ca2+ uptake rate if non-energized mitochondria are first exposed for different periods of time to micromolar Ca2+ concentrations. A simple experiment demonstrating this phenomenon in isolated mitochondria is shown in Figure 1. Non-respiring mitochondria are exposed to 3 μM Ca2+ for 30 s, 1 or 7 min before Ca2+ uptake is triggered by the addition of succinate. The longer the incubation in 3 μM Ca2+, the faster the initial rate of Ca2+ uptake. This phenomenon is inhibited by the calmodulin inhibitor calmidazolium, but it is insensitive to CAM kinase II inhibitors.7 The molecular mechanisms involved in this phenomenon remain mysterious, but it should be taken into account to explain some of the kinetic behavior of mitochondria in living cells.

Time dependence of the Ca2+ activation of mitochondrial Ca2+ uptake. The experiment was carried out in isolated mitochondria incubated in sucrose medium, essentially as described in Pozzan et al.2 Extramitochondrial Ca2+ was measured with the fluorescent Ca2+ indicator Calcium Green-5N. (a) Non-respiring mitochondria (treated with rotenone and oligomycin) were incubated in medium containing 3 μM CaCl2 for different periods of time (30 s, 1 and 7 min) before adding 2 mM succinate to initiate Ca2+ uptake. Typical traces are shown. (b) Initial rate of mitochondrial Ca2+ uptake as a function of the preincubation time in 3 μM CaCl2; each point is the mean of two experiments carried out in the same mitochondrial preparation. Similar results were obtained in at least six independent trials
The Ca2+ efflux mechanisms appear to be regulated by the Ca2+, Na+ (or H+) concentrations outside and/or inside the mitochondria and by in vivo treatment with glucagon and β-adrenergic agonists (for a review, see Rizzuto et al.8). The Na+/Ca2+antiporters are most likely electrogenic9 (three Na+/H+ ions are exchanged for one Ca2+) and, accordingly, a negative membrane potential inside favors Ca2+ extrusion. Ca2+ efflux can also occur through the uniporter if the membrane potential is collapsed. Of interest, the classical inhibitor of the uniporter, Ruthenium Red (RR), poorly inhibits the Ca2+ release, which was largely incomprehensible until it was elucidated by the channel nature of the uniporter10 and see below: RR, a cation with six positive charges, is attracted into the Ca2+ channel at negative membrane potential in the matrix (probably occluding it), but is released when the membrane potential is collapsed. Another Ca2+ efflux pathway is the so-called permeability transition pore, PTP (see below). Indeed, as a consequence of the collapse of membrane potential caused by the PTP activation, Ca2+ can be lost both through the open PTP itself and through reversal of the uniporter.
All the functional characteristics of mitochondrial Ca2+ handling machinery described so far were largely known by the end of the 1970s. At the time, mitochondria were believed to represent an important Ca2+ storage organelle, from where Ca2+ could be mobilized upon cell activation. The demonstration that mitochondria in healthy living cells contain trace amounts of Ca2+ and the characterization of the uniporter kinetics mentioned above (low influx rate at the physiological Ca2+ concentrations not only of resting cells, but also upon activation), convinced most investigators that mitochondrial Ca2+ uptake played a marginal role in the overall control of Ca2+ homeostasis under physiological conditions and that it becomes important only under frank pathological situations.11 As to the latter (see below), it was in fact already well known that necrotic cells had often electron-dense material within mitochondria, represented by precipitates of Ca2+ phosphate.11 As a consequence, during the decade that followed, the scientific community paid little attention to mitochondrial Ca2+ handling; episodically, however, papers were published that indirectly suggested that this organelle could have a more important role in the cell physiology of Ca2+ handling (for a review, see Pozzan et al.12).
A major change happened in the early 1990s, when we pioneered the use of recombinant targeted aequorins to monitor directly [Ca2+]m.13 With this novel methodology we could show that a rise in [Ca2+]m occurred for any, even very small, change in [Ca2+]c (as predicted by the model discussed above); at the same time, and unexpectedly, we also observed that in a large number of cells a physiological rise of [Ca2+]c was accompanied by very fast and large increases in [Ca2+]m – much faster and much larger than the prediction based on the low affinity of the uniporter in vitro.14, 15 This new paradox was unraveled through the hypothesis of local microdomains. The fast Ca2+ uptake observed depends on the generation of very high [Ca2+], close to mitochondria, where the organelles happen to be closely apposed to Ca2+ channels (plasma membrane or ER/SR); it is these microdomains of high [Ca2+] that induce a very fast, but transient accumulation of Ca2+ in the mitochondria14 (for a recent review, see Rizzuto and Pozzan3). The hypothesis has received broad experimental support, leading to a renewed interest in the mechanisms and role of mitochondrial Ca2+ handling. The observation that really brought mitochondria back into the limelight was published only a few years later: the demonstration that programmed cell death could depend in many instances on the release from mitochondria of a small protein, cytochrome c, located between the inner and the outer mitochondrial membranes, and that this release could in turn lead to the activation of effector caspases (for a review, see Ferri and Kroemer16). The link between cytochrome c release, apoptosis and mitochondrial Ca2+ was immediately clear: overaccumulation of Ca2+ by mitochondria was already known to activate the PTP (in a cyclosporine A sensitive mechanisms, see below) and this large membrane pore in the inner mitochondrial membrane can lead to matrix swelling, causing the rupture of the outer mitochondrial membrane and release of cytochrome c.16, 17, 18
Mitochondrial Ca2+ handling thus became the center of a very important biological problem, with key pathophysiological consequences for a number of human pathologies, from cell death due to ischemia, to excitotoxicity in the CNS, to degenerative diseases. Add to this the finding that pro- and anti-apoptotic genes can modulate mitochondrial Ca2+ handling (see below), and the importance of mitochondrial Ca2+ handling properties becomes immediately clear, with excursions even into problems related to cell growth and cancer development.
It should be stressed that, as far as mitochondrial Ca2+ handling is concerned, extreme care should be taken in comparing results obtained in isolated organelles with those within intact cells. The isolation of the organelles disrupts the anatomical connections with other cellular structures, in particular, the plasma membrane and ER/SR Ca2+ channels, that are vital for the generation of Ca2+ microdomains.3 Within living cells, mitochondria are exposed both to oxidizable substrates and to a high concentration of ATP, generated by the organelles themselves and by glycolysis. This latter, trivial consideration, is often overlooked by many investigators. Thus, if isolated mitochondria are incubated with a respiratory substrate (e.g. glutamate) the inhibition of the respiratory chain (by rotenone or antimycin) prevents mitochondrial Ca2+ accumulation because the membrane potential generated by the respiratory chain is rapidly collapsed. On the contrary, in the majority of living cells, Ca2+ accumulation in response to a stimulus is practically unaffected by the same drugs, because the membrane potential can be regenerated by reversal of the mitochondrial ATPase, using ATP produced by glycolysis. To block efficiently mitochondrial Ca2+ uptake in a living cell one must use either an uncoupler (but other side effects need to be taken into consideration) or a combination of a respiratory chain blocker and oligomycin, to block the ATPase as well. Effects on Ca2+ handling, often reported, of respiratory chain inhibitors only (or oligomycin alone) most likely reflect the consequences of these drugs on functions that are not linked to mitochondrial Ca2+ uptake.
Ironically, in spite of the enormous interest in mitochondrial Ca2+ homeostasis, the molecular identification of the players involved is still missing. The search for the mitochondrial Ca2+ uniporter, for example, has been a frustrating issue for the last 40 years. Several groups claimed to have isolated this molecular complex, but none of these claims has been ultimately confirmed. At one stage, the best known candidate was the so-called ‘P30 glycoprotein’, a peripheral glycosylated membrane protein that the authors thought could transport Ca2+ across the inner membrane with a mechanism similar to that of classical antibiotic ionophores, such as valinomycin.19, 20 In 1979, based on a series of indirect data, we proposed that the uniporter was a gated ion channel21 that is an integral protein of the inner mitochondrial membrane. This proposal was never followed up until Clapham and coworkers, by patch clamping the inner mitochondrial membrane, very elegantly showed that the uniporter is indeed a selective divalent cation channel.10
The search for the antiporters has been less intense, and we are unaware of any conclusive study leading to their identification. To our knowledge, in silico searches for potential candidate genes, have fared no better – in spite of the fact that the identified members of the mitochondrial proteome continue to grow steadily. In summary, over 40 years after the initial functional identification of the mitochondrial Ca2+ uniporter (and over 30 after that of the antiporters) the molecular identities of these key regulators of the mitochondrial Ca2+ handling machinery remain unknown.
Physiological Roles of Mitochondrial Ca2+ Accumulation
What is the function of mitochondrial Ca2+ uptake? It is beyond the focus of this review to discuss the physiological consequences of mitochondrial Ca2+ uptake in detail. A few key physiological processes – directly or indirectly modulated by mitochondrial Ca2+ uptake and release – must, however, be addressed briefly.
One of the best characterized functions of mitochondrial Ca2+ uptake is the control of organelle metabolic activity. Three crucial metabolic enzymes within the matrix (pyruvate, α-ketoglutarate and isocitrate dehydrogenases) are activated by Ca2+, using two distinct mechanisms. In the case of pyruvate dehydrogenase, a Ca2+-dependent dephosphorylation step is involved; in the other two cases, the activation is through direct binding of Ca2+ to the enzyme complex (for a recent review, see Rizzuto and Pozzan3). Given that these enzymes represent the rate limiting step for feeding electrons into the respiratory chain, Ca2+ within the matrix is ultimately the positive modulator of mitochondrial ATP synthesis; this aspect was directly addressed by Rizzuto's group a few years ago using targeted recombinant luciferase to monitor, in living cells, the ATP concentration within the cytoplasm and the mitochondrial matrix.22 Recently, also some metabolite transporters have been shown to be regulated by Ca2+ and to participate in the enhancement of aerobic metabolism upon cell activation.3 One issue remains to be clarified: given that the matrix dehydrogenases are activated by [Ca2+] in the low micromolar range, why do [Ca2+]m rises reach values up to several tens or hundreds of μM? Several explanations could be offered. The first is that most [Ca2+]m estimates were obtained in prolonged, supramaximal stimulations with Ca2+-mobilizing agonists, and it is likely that these conditions do not fully mimic the response to physiological challenges (that give rise to smaller and more transient [Ca2+]c increases). The second is that a large [Ca2+]m response (together with other mechanisms, such as the enzymatic delay in the rephosphorylation of the PDH complex) could be a route to extend the metabolic activation well beyond the duration of the [Ca2+]c rise. The latter explanation may be particularly relevant in heart cells, where [Ca2+]m oscillates on a beat-to-beat basis, while the activity of the dehydrogenases must be constantly elevated.
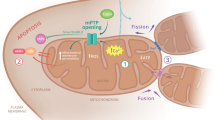
A second process that depends on mitochondrial Ca2+ handling regards the kinetics of cytosolic Ca2+ changes in response to agonist stimulation. Specific examples in this case are too numerous to be addressed in detail here, but the following aspects provide a general framework. Mitochondria, by buffering local [Ca2+] (generated by Ca2+ channels on the plasma membrane or the ER/SR), can augment or decrease the release/influx of Ca2+ and modulate the frequency of Ca2+ oscillations; this phenomenon depends on the cell type and the channel involved (see, e.g., Rizzuto et al.,23 Collins et al.,24 Hajnóczky et al.,25 Landolfi et al.,26 Vay et al.27). In addition, mitochondria can also exert a more classical buffering role. One example is the cluster of mitochondria that isolate functionally distinct domains in polarized cells: a mitochondrial ‘firewall’ was shown to prevent the spread of Ca2+ signals from the apical region of pancreatic acinar cell to the basolateral region.28 Another example, this time in neurons, sees mitochondria buffering [Ca2+] increases in defined cellular regions, that is the presynaptic motoneuron ending.29 The mitochondrial Ca2+ efflux mechanisms have also been involved in shaping the cytoplasmic Ca2+ kinetics in neurons in response to intense electrical stimulation (by slowly releasing the accumulated Ca2+),30 or by allowing the efficient refilling of the ER and thus modulating Ca2+ oscillations.27, 31 While the above mentioned roles are more or less similar in every cell type, there are also other roles of mitochondrial Ca2+ that appear to be tissue specific. The two examples best studied are the endocrine pancreas, where mitochondrial Ca2+ modulates insulin secretion,32 and the granulosa cells of the adrenal gland, where it controls a key step in aldosterone synthesis.33 A schematic view of mitochondrial Ca2+ regulation and of the physiological processes that are modulated is presented in Figure 2.

Schematic view of the process of Ca2+ homeostasis in mitochondria. The key molecular players in the process of Ca2+ accumulation/release are schematically drawn with their supposed intramitochondrial localization. ETC, electron transport chain, VDAC, voltage-dependent anion channel, OMM: outer mitochondrial membrane, IMM, inner mitochondrial membrane, PTP, permeability transition pore
Currently, the most studied role of mitochondrial Ca2+ handling is, by far, the control of apoptosis/necrosis. Regarding apoptosis, it is probably a semantic problem to include it in the physiology or pathology of the cell. For reasons of simplicity we have decided to discuss apoptosis in the next chapter.
Mitochondrial Ca2+ Homeostasis and Cell Death by Necrosis and Apoptosis
Necrosis is the catastrophic derangement of cell integrity and function that follows exposure to different types of cell injury, leading to the activation of Ca2+-activated hydrolysing enzymes.34, 35 The fact that major increases in [Ca2+]c are capable of causing necrosis has been known for a long time. Similarly, it is well established that mitochondria represent a major target of Ca2+-dependent necrosis, as demonstrated by the formation of the Ca2+-phosphate deposits in necrotic cells, mitochondrial membrane potential collapse, rapid drop in ATP levels and production of ROS (for a review, see Rizzuto et al.36). More recently, it has been shown that Ca2+ can also play a central role in triggering some forms of a more subtle and controlled pathway of cell death, i.e. the apoptotic program. In this case, what attracted the attention of many scientists to Ca2+ was the discovery of the effects of Bcl-2 on Ca2+ signaling. The first of such observations dates back to 1993, when it was found that Bcl-2 overexpression decreases the amount of Ca2+ capable of being mobilized from the ER.37 In 2000, an additional seminal observation was made contemporarily by our group and by that of K.H. Krause:38, 39 briefly, it was not only found that cells overexpressing Bcl-2 display an enhanced survival upon treatment with some apoptogenic drugs (as expected), but also, surprisingly, that such cells display an ∼30% reduction in the [Ca2+] levels within the lumen of the ER (and of the Golgi apparatus). Both groups also showed that the major consequence of this reduction in stored Ca2+ was a large decrease of [Ca2+]c and [Ca2+]m increases elicited in these cells by stimuli coupled to IP3 generation.38, 39 The question then arose as to whether this reduction of [Ca2+] within the stores, due to overexpression of anti-apoptotic proteins, is relevant for apoptosis or whether is it a side effect of the overexpression of these proteins. Indirect support for the first hypothesis came from the work of Pinton et al.40 which showed that the reduction of [Ca2+] within the stores plays an important role in the anti-apoptotic mechanism of Bcl-2: mimicking the Bcl-2 effect on [Ca2+] by different pharmacological and molecular approaches (but in the absence of the oncoprotein) the cells were protected from the apoptotic stimulus ceramide. Of interest, treatments that increased [Ca2+] within the stores had the opposite effect on the susceptibility of cells to the apoptotic stimulus.40
These observations fitted nicely with other data published in the same period, totally independently, by other groups, such as: (i) the association of Bcl-2 with mitochondrial and ER membranes;41 (ii) the demonstration, mentioned above, that excess Ca2+ accumulation by mitochondria leads to opening of the PTP and to massive mitochondrial swelling;17, 18, 42 and (iii) that cytochrome c is released into the cytosol in response to several apoptotic stimuli. Taken together these data allowed the formulation of a rational working hypothesis: (i) an apoptotic insult causes the release of Ca2+ from the ER; (ii) the amount of Ca2+ content of the ER determines the amount of Ca2+ taken up by mitochondria; (iii) when the latter organelles accumulate an excess of Ca2+ the PTP is activated, release of cytochrome c and other pro-apoptotic factors occur and eventually executor caspases are irreversibly activated. It could be argued that this model is in contradiction with the well established fact that similarly large (or even larger) Ca2+ releases and mitochondrial accumulations, elicited by a number of stimuli, do not trigger cell death, but rather are beneficial, for example by increasing the cellular ATP levels. Pinton et al.,40 to solve this contradiction, proposed the ‘double hit’ hypothesis; that is that apoptotic stimuli, such as ceramide, have a dual target: on the one hand cause the release of Ca2+ from the ER and its uptake by mitochondria, on the other make the mitochondria more sensitive to the potential Ca2+ damaging effects. This hypothesis fits nicely with elegant studies by Hajnoczky and coworkers. In particular, they showed that ceramide facilitates PTP opening, thus transforming physiological IP3-mediated Ca2+ signals into inducers of apoptosis.43 Finally, unpublished results by our group show that activators of mitochondrial Ca2+ uptake potently synergize with sub-threshold doses of apoptotic agents.
This model of Ca2+-dependent apoptosis triggering was subsequently supported by a series of observations, the most important of which can be briefly described as follows. Scorrano et al. demonstrated not only that embryonic fibroblasts from knockout mice lacking the pro-apoptotic proteins Bax and Bak are very resistant to apoptotic death, but also that they have a dramatic reduction in the [Ca2+] within the ER and a drastic reduction in the transfer of Ca2+ from the ER to mitochondria.44 Furthermore, silencing Bcl-2 in Bax/Bak knockout cells partially restores [Ca2+] values within the ER to control levels44 and normalizes the mitochondrial Ca2+ responses. In the Bax/Bak knockout cells, when the ER Ca2+ levels were restored by recombinantly over-expressing the ER Ca2+ ATPase (SERCA2b), not only was mitochondrial Ca2+ uptake in response to stimulation re-established, but the cells regained sensitivity to apoptotic stimuli such as arachidonic acid, C2-ceramide and oxidative stress.44 Along the same line of reasoning, calreticulin over-expressing cells, that have an augmented ER Ca2+ content, are more susceptible to apoptosis induced by ceramide treatment,40 while calreticulin knockout cell lines, that show a marked decrease in ER Ca2+ release upon cell stimulation, are more resistant to apoptosis.45 Chami et al.46 also showed that early after overexpression of Bax in HeLa cells the [Ca2+] of the ER is higher than in controls. Finally, Tsien and coworkers not only confirmed that Bcl-2 overexpression leads to decreased ER Ca2+ levels, but also showed that the green tea compound epigallocatechin gallate, known to bind and inactivate Bcl-2, restored [Ca2+] of the ER to that of normal cells.47 We are aware that not all experts in the field concur with these conclusions – see, for example, Chen et al.,48 He et al.,49 Lam et al.,50 Wang et al.,51, 52 Zhong et al.,53 Ichimiya et al.,54 Wei et al.,55 Kuo et al.,56 Zhu et al.57 But it is our biased opinion that in many of these studies the different conclusions depend on the specific experimental approach (indirect methods of monitoring Ca2+ in the ER, use of clones, etc.). A very interesting possibility would be that the modulation of Ca2+ handling by pro- and anti-apoptotic proteins is exerted because they can alter the gating properties of specific isoform of the IP3 receptor, IP3R. If this is the case the effects of the pro- and anti-apoptotic proteins on Ca2+ handling should depend on the specific cell model employed and the expression profile of IP3Rs. Recent evidence indicates that indeed the last hypothesis may be true.58, 59, 60 An obvious corollary to this hypothesis is that the effect of pro- and anti-apoptotic proteins is only one of the mechanisms through which these proteins control cell death, as indeed clearly shown by Scorrano et al.44
In summary, Bcl-2 and other anti-apoptotic proteins reduce ER Ca2+ levels, and consequently moderate the efficacy of apoptotic mediators that use Ca2+ signals (and the involvement of mitochondria as downstream effectors) as a potentiation/commitment factor. Conversely, Bax (and other pro-apoptotic proteins of the family) enhances the loading of the ER Ca2+ store, and thus boosts the Ca2+ load to which the apoptotic effector systems (including mitochondria) are exposed upon physiological and/or pathological challenges. The model is schematically described in Figure 3.

Proposed model of the interplay among mitochondria, ER and pro- or anti-apoptotic Bcl-2 family members in triggering Ca2+-modulated apoptosis
Ca2+ and Human Genetic Diseases
The number of clinically relevant pathological events in which the proposed model of Ca2+ activated cell death may play a central role are numerous: for example, ischemic death in the heart and other tissues, glutamate dependent excitotoxicity in the CNS, Parkinson's disease, Alzheimer's disease, and even Ullrich muscular dystrophy (UMD) and Duchenne muscular dystrophy.
In this last chapter we will briefly discuss the involvement of Ca2+ and mitochondria in UMD and Alzheimer's disease, given that in these latter two pathologies the role of Ca2+ and of mitochondria is less obvious and/or still a matter of some controversy.
UMD is a severe muscular dystrophy, with different age of onset and gravity. The involvement of mitochondria in the pathogenesis of the fiber degeneration in UMD was a very surprising and unexpected observation. UMD, and the less severe Bethlem myopathy, are muscle diseases caused by defects in collagen VI. Bernardi and coworkers,61 showed that in muscle fibers of knockout mice for collagen VI, mitochondria have a latent defect, that is a strong susceptibility of the PTP to open for mild mitochondrial insults; mitochondria isolated from knockout animals in vitro have a very modest capacity to accumulate Ca2+ in response to Ca2+ challenges, due to very rapid PTP opening (sensitive to cyclosporine A). The molecular link between the collagen VI defect and the mitochondrial instability is still largely mysterious. From the therapeutical point of view, the important fact is that injection in the knockout mouse of the PTP inhibitor cyclosporine A led to a dramatic recovery of the muscle lesions. Even more important, the same group very recently showed in biopsies from human patients that chelation of intracellular Ca2+, addition of collagen VI or treatment with cyclosporins that inhibit the PTP could, in vitro, ameliorate mitochondrial dysfunctions of myoblasts from UMD patients,62 thus offering for the first time the possibility of a pharmacological therapy for this incurable and devastating genetic disease.
Recent evidence suggests that Ca2+ may play a key role also in Alzheimer's disease (AD), or at least in the genetic forms of the disease. AD is the most common neurodegenerative disorder63 and it accounts for about 50% of the cases of senile dementia. This pathology, first described by Alois Alzheimer in 1906, is characterized by cortical atrophy, accumulation of abnormal fibres in neuronal cell bodies, and the presence, in the extracellular space, of senile plaques, whose main component is the so-called Aβ peptide. The latter derives from the transmembrane protein amyloid precursor protein (APP), which can be alternatively processed by three different enzymes, named α, β, and γ secretases. The combined action of β and γ secretases leads to the formation of a soluble fragment (sAPPβ) and of the Aβ peptide, together with its cytosolic counterpart AICD (APP IntraCellular Domain).
Mutations in the genes encoding for APP, Presenilin1 and 2 (PS1 and PS2), two proteins belonging to the γ-secretase enzymatic complex, have been linked to the familial form of AD (FAD; for a recent review, see St George-Hyslop and Petit64). Since the common phenotype of all these mutations is an increased Aβ production, Hardy et al.65 proposed the amyloid cascade hypothesis, stating that accumulation of Aβ is the chief molecular event that causes the onset of the disease (but see also De Strooper66).
About 10 years ago it has been proposed that an alteration in intracellular Ca2+ homeostasis could contribute to the development of FAD. The majority of published data, obtained mainly by analyzing PS1 mutations, report that mutated presenilins increase the ER Ca2+ content (for a recent review, see Smith et al.67). The hypothesis was thus proposed68 that a Ca2+ overload could either lead to an increased Aβ production or, alternatively, that the overload of Ca2+ stores caused by presenilin mutations could exacerbate the Aβ toxicity by favoring Ca2+-dependent cell death. A strong support for this hypothesis came recently from the demonstration that wild type presenilins, but not the mutated forms, can form leak channels in the ER (they also form divalent cation permeable channels in lipid bilayers69). According to the above discussed model of ER-mitochondria crosstalk in triggering apoptosis, it could be speculated that in neurons expressing the mutated PS isoforms a reduction in the ER Ca2+ leak leads to over-accumulation of Ca2+ in the ER and thus can favor its transfer to mitochondria, leading to neuronal apoptosis. Although this hypothesis has gained popularity, there are data that are difficult to reconcile with it. In particular, Zatti et al. 70, 71, 72 showed that some FAD-linked PS2 mutations caused a reduction, yet not an increase, in ER Ca2+ levels.
A possible alternative hypothesis could be suggested for hereditary AD due to presenilin mutations: by increasing Aβ formation (and in particular of the 42 AA form), the mutated enzymes lead to neuronal degeneration. At the same time, by modulating Ca2+ within the ER they can exacerbate or reduce the toxicity of Aβ. Presenilin mutations (PS1 in particular) that cause an increase in the ER Ca2+ exacerbate cell death, while mutations (in particular of PS2) that decrease the ER Ca2+ levels partially protect cells from Ca2+-dependent cell death. This hypothesis would be consistent with the afore mentioned role of ER and mitochondrial Ca2+ relationship, and with the clinical observation that FAD-linked PS2 mutations have been associated to milder phenotypes.71, 72
In conclusion, the capacity of mitochondria to accumulate and release Ca2+ appears intimately linked to the multiple roles of these organelles within cells: on the one hand, Ca2+ accumulation/release is instrumental in modulating the key bioenergetic role of mitochondria (e.g., respiratory substrate oxidation and ATP synthesis) and in modulating the kinetics and amplitude of the [Ca2+]c signal (and thus the cell functions that depend on this second messenger); on the other, massive accumulation of Ca2+ in the mitochondria leads to necrotic cells death; finally more modest increases in [Ca2+]m, but in the presence of other toxic insults, trigger the mitochondrial gateway to apoptosis.
Abbreviations
- UMD:
-
ullrich muscular dystrophy
- APP:
-
amyloid precursor protein
- AICD:
-
APP intracellular domain
- AD:
-
Alzheimer's disease
References
Vasington F, Murphy JV . Ca2+ uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem 1962; 237: 2670–2677.
Pozzan T, Bragadin M, Azzone GF . Disequilibrium between steady-state Ca2+ accumulation ratio and membrane potential in mitochondria. Pathway and role of Ca2+ efflux. Biochemistry 1977; 16: 5618–5625.
Rizzuto R, Pozzan T . Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 2006; 86: 369–408.
Montero M, Lobatón CD, Moreno A, Alvarez J . A novel regulatory mechanism of the mitochondrial Ca2+ uniporter revealed by the p38 mitogen-activated protein kinase inhibitor SB202190. FASEB J 2002; 16: 1955–1957.
Pinton P, Leo S, Wieckowski MR, Di Benedetto G, Rizzuto R . Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J Cell Biol 2004; 165: 223–232.
Kroner H . Allosteric regulation' of calcium-uptake in rat liver mitochondria. Biol Chem Hoppe Seyler 1986; 367: 483–493.
Moreau B, Nelson C, Parekh AB . Biphasic regulation of mitochondrial Ca2+ uptake by cytosolic Ca2+ concentration. Curr Biol 2006; 16: 1672–1677.
Rizzuto R, Bernardi P, Pozzan T . Mitochondria as all-round players of the calcium game. J Physiol 2000; 529 Pt 1 37–47.
Jung DW, Baysal K, Brierley GP . The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem 1995; 270: 672–678.
Kirichok Y, Krapivinsky G, Clapham DE . The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004; 427: 360–364.
Carafoli E . Intracellular calcium homeostasis. Annu Rev Biochem 1987; 56: 395–433.
Pozzan T, Magalhães P, Rizzuto R . The comeback of mitochondria to calcium signalling. Cell Calcium 2000; 28: 279–283.
Rizzuto R, Simpson AWM, Brini M, Pozzan T . Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992; 358: 325–327.
Rizzuto R, Brini M, Murgia M, Pozzan T . Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993; 262: 744–747.
Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T . Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol 1994; 126: 1183–1194.
Ferri KF, Kroemer G . Organelle-specific initiation of cell death pathways. Nat Cell Biol 2001; 3: E255–E263.
Zamzami N, Kroemer G . The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2001; 2: 67–71.
Kroemer G, Zamzami N, Susin SA . Mitochondrial control of apoptosis. Immunol Today 1997; 18: 44–51.
Saris NE, Sirota TV, Virtanen I, Niva K, Penttila T, Dolgachova LP et al. Inhibition of the mitochondrial calcium uniporter by antibodies against a 40-kDa glycoproteinT. J Bioenerg Biomembr 1993; 25: 307–312.
Panfili E, Sandri G, Sottocasa GL, Lunazzi G, Liut G, Graziosi G . Specific inhibition of mitochondrial Ca2+ transport by antibodies directed to the Ca2+-binding glycoprotein. Nature 1976; 264: 185–186.
Bragadin M, Pozzan T, Azzone GF . Activation energies and enthalpies during Ca2+ transport in rat liver mitochondria. FEBS Lett 1979; 104: 347–351.
Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R . Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA 1999; 96: 13807–13812.
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998; 280: 1763–1766.
Collins TJ, Lipp P, Berridge MJ, Bootman MD . Mitochondrial Ca2+ uptake depends on the spatial and temporal profile of cytosolic Ca2+ signals. J Biol Chem 2001; 276: 26411–26420.
Hajnóczky G, Csordás G, Madesh M, Pacher P . The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. J Physiol 2000; 529 (Pt 1) 69–81.
Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM . Ca2+ homeostasis in the agonist-sensitive internal store: Functional interactions between mitochondria and the ER measured in situ in intact cells. J Cell Biol 1998; 142: 1235–1243.
Vay L, Hernandez-Sanmiguel E, Santodomingo J, Lobaton CD, Moreno A, Montero M et al. Modulation of Ca2+-release and Ca2+ oscillations by mitochondrial Ca2+ uniporter stimulation. J Physiol 2007 (Published online, at 10.1113/j physiol.2006.126391).
Park MK, Ashby MC, Erdemli G, Petersen OH, Tepikin AV . Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J 2001; 20: 1863–1874.
David G, Barrett JN, Barrett EF . Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol 1998; 509: 59–65.
Colegrove SL, Albrecht MA, Friel DD . Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons. Reconstruction of the recovery after depolarization-evoked [Ca2+]i elevations. Journal of General Physiology 2000; 115: 371–388.
Ishii K, Hirose K, Iino M . Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep 2006; 7: 390–396.
Maechler P, Kennedy ED, Pozzan T, Wollheim CB . Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO J 1997; 16: 3833–3841.
Brandenburger Y, Kennedy ED, Python CP, Rossier MF, Vallotton MB, Wollheim CB et al. Possible role for mitochondrial calcium in angiotensin II- and potassium-stimulated steroidogenesis in bovine adrenal glomerulosa cells. Endocrinology 1996; 137: 5544–5551.
Nicotera P, Orrenius S . The role of calcium in apoptosis. Cell Calcium 1998; 23: 173–180.
Budd SL, Nicholls DG . A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem 1996; 66: 403–411.
Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhães PJ et al. Calcium and apoptosis: facts and hypotheses. Oncogene 2003; 22: 8619–8627.
Baffy G, Miyashita T, Williamson JR, Reed JC . Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3- dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem 1993; 268: 6511–6519.
Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T et al. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J Cell Biol 2000; 148: 857–862.
Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP et al. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA 2000; 97: 5723–5728.
Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R . The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 2001; 20: 2690–2701.
Lithgow T, van Driel R, Bertram JF, Strasser A . The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ 1994; 5: 411–417.
Bernardi P . Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 1999; 79: 1127–1155.
Szalai G, Krishnamurthy R, Hajnoczky G . Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J 1999; 18: 6349–6361.
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003; 300: 135–139.
Nakamura K, Bossy-Wetzel E, Burns K, Fadel MP, Lozyk M, Goping IS et al. Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J Cell Biol 2000; 150: 731–740.
Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC et al. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem 2004; 279: 54581–54589.
Palmer AE, Jin C, Reed JC, Tsien RY . Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA 2004; 101: 17404–17409.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166: 193–203.
He H, Lam M, McCormick TS, Distelhorst CW . Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol 1997; 138: 1219–1228.
Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW . Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci USA 1994; 91: 6569–6573.
Wang CT, Grishanin R, Earles CA, Chang PY, Martin TF, Chapman ER et al. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science 2001; 294: 1111–1115.
Wang H-G, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999; 284: 339–343.
Zhong F, Davis MC, McColl KS, Distelhorst CW . Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol 2006; 172: 127–137.
Ichimiya M, Chang SH, Liu H, Berezesky IK, Trump BF, Amstad PA . Effect of Bcl-2 on oxidant-induced cell death and intracellular Ca2+ mobilization. Am J Physiol 1998; 275: C832–C839.
Wei H, Wei W, Bredesen DE, Perry DC . Bcl-2 protects against apoptosis in neuronal cell line caused by thapsigargin-induced depletion of intracellular calcium stores. J Neurochem 1998; 70: 2305–2314.
Kuo TH, Kim H-RC, Zhu L, Yu Y, Lin H-M, Tsang W . Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene 1998; 17: 1903–1910.
Zhu L, Ling S, Yu XD, Venkatesh LK, Subramanian T, Chinnadurai G et al. Modulation of mitochondrial Ca(2+) homeostasis by Bcl-2. J Biol Chem 1999; 274: 33267–33273.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA 2005; 102: 105–110.
Hanson CJ, Bootman MD, Roderick HL . Cell signalling: IP3 receptors channel calcium into cell death. Curr Biol 2004; 14: R933–R935.
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB et al. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 2005; 7: 1021–1028.
Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 2003; 35: 367–371.
Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci USA 2007; 104: 991–996.
Goedert M, Spillantini MG . A century of Alzheimer's disease. Science 2006; 314: 777–781.
St George-Hyslop PH, Petit A . Molecular biology and genetics of Alzheimer's disease. C R Biol 2005; 328: 119–130.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
De Strooper B . Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 2007; 8: 141–146.
Smith IF, Green KN, LaFerla FM . Calcium dysregulation in Alzheimer's disease: Recent advances gained from genetically modified animals. Cell Calcium 2005; 38: 427–437.
LaFerla FM . Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci 2002; 3: 862–872.
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell 2006; 126: 981–993.
Zatti G, Ghidoni R, Barbiero L, Binetti G, Pozzan T, Fasolato C et al. The presenilin 2 M239I mutation associated with familial Alzheimer's disease reduces Ca2+ release from intracellular stores. Neurobiol Disease 2004; 15: 269–278.
Giacomello M, Barbiero L, Zatti G, Squitti R, Binetti G, Pozzan T et al. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer's disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol Disease 2005; 18: 638–648.
Zatti G, Burgo A, Giacomello M, Barbiero L, Ghidoni R, Sinigaglia G et al. Presenilin mutations linked to familial Alzheimer's disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006; 39: 539–550.
Acknowledgements
The original work of the authors was supported by grants from Telethon, AIRC, the Italian Ministry of University, the Veneto region program ‘Biotech II’ and by a grant from the University of Padua to PP.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by P Nicotera
Rights and permissions
About this article
Cite this article
Giacomello, M., Drago, I., Pizzo, P. et al. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ 14, 1267–1274 (2007). https://doi.org/10.1038/sj.cdd.4402147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402147
Keywords
This article is cited by
-
Superparamagnetic Iron Oxide Nanoparticles Induce Apoptosis in HT-29 Cells by Stimulating Oxidative Stress and Damaging DNA
Biological Trace Element Research (2023)
-
Characterization of oxidation of glutathione by cytochrome c
Journal of Bioenergetics and Biomembranes (2022)
-
The multiple mechanisms of MCL1 in the regulation of cell fate
Communications Biology (2021)
-
Calcium dysregulation mediates mitochondrial and neurite outgrowth abnormalities in SOD2 deficient embryonic cerebral cortical neurons
Cell Death & Differentiation (2019)
-
Mitochondria damaged by Oxygen Glucose Deprivation can be Restored through Activation of the PI3K/Akt Pathway and Inhibition of Calcium Influx by Amlodipine Camsylate
Scientific Reports (2019)
