
Abstract
Dexamethasone (DEX) pretreatment protected hepatocytes from TNF-α plus actinomycin D (ActD)-induced apoptosis by suppressing caspase-8 activation and the mitochondria-dependent apoptosis pathway. DEX treatment upregulated cellular FLICE inhibitory protein (cFLIP) expression, but did not alter the protein levels of Bcl-2, Bcl-xL, Mcl-1, and cIAP as well as Akt activation. The increased cFLIP mRNA level by DEX was inhibited by ActD, indicating that DEX upregulates cFLIP expression at the transcriptional step. DEX also inhibited Jo2-mediated hepatocyte apoptosis by blocking the formation of the death-inducing signaling complex and caspase-8 activation. Specific downregulation of cFLIP expression using siRNA reversed the antiapoptotic effect of DEX by increasing caspase-8 activation. Moreover, DEX administration into mice increased cFLIP expression in the liver and prevented Jo2-induced hepatic injury by inhibiting caspase-8 and -3 activities. Our results indicate that DEX exerts a protective role in death receptor-induced in vitro and in vivo hepatocyte apoptosis by upregulating cFLIP expression.
Similar content being viewed by others


Introduction
Apoptosis, known as programmed cell death, is a cellular self-destruction mechanism involved in a variety of biological events such as developmental remodeling, tissue homeostasis, and the removal of unwanted cells in most living tissues, including the mammalian liver. Similar to other organs, apoptosis plays a role in the pathogenesis of fulminant hepatic failure, hepatitis, organ dysfunction after liver transplantation, and liver ischemia/reperfusion injury.1 Hepatocyte apoptosis can be initiated by exposure to toxic substances or ligation of members of the death receptor family including tumor necrosis factor-alpha (TNF-α) receptor (TNF-R) and Fas.2
Activation of TNF-R and Fas elicits the recruitment of a cellular adaptor protein, Fas-associated death domain-containing protein (FADD), to the intracellular death domain of the activated receptors.3 FADD also recruits procaspase-8 and forms the death-inducing signaling complex (DISC), leading to the activation of procaspase-8 by proteolytic processing. Active caspase-8 cleaves p22 Bid at Asp59 into the truncated p15, which translocates into the mitochondrial membrane and induces cytochrome c release. The accumulation of cytochrome c in the cytosol results in the formation of the apoptosome via interaction with Apaf-1, procaspase-9, and dATP and leads to caspase-9 activation. Activated caspase-9 subsequently activates the executioner proteases including caspase-3, which in turn liberates a DNase termed CAD (caspase-activated DNase) from an inhibitor of CAD (ICAD/DFF-45) by cleaving the ICAD protein.4 This process leads to DNA degradation, a hallmark event in apoptosis.
Dexamethasone (DEX), a synthetic glucocorticoid, has been used as a potent immunesuppressive therapeutic agent for several inflammatory diseases, as it suppresses the expression of inflammatory genes including TNF-α, cyclooxygenase-2, and inducible nitric oxide synthase via the inhibition of NF-κB activation.5 Furthermore, DEX has been shown to either inhibit apoptosis in some cells such as human neutrophils,6 tumor cells,7 and primary human and rat hepatocytes8 or induce apoptosis in others including thymocytes and lymphocytes.9, 10 The antiapoptotic effect by DEX is regulated by several sets of genes, the best characterized of which are the Bcl-2 family such as Bcl-2 and Bcl-xL, which prevent spontaneous apoptosis of cultured primary hepatocytes.11 It is also known that DEX abolishes TNF-α-induced cytoxicity of several tumor cell lines via inhibition of phospholipase A2 activity.12, 13 However, the mechanism behind the antiapoptotic effects of DEX has rarely been investigated in death receptor-mediated hepatocyte apoptosis.
We here determined the molecular mechanism by which DEX protects human and rat hepatocytes from death receptor-mediated apoptosis. We found that DEX inhibits apoptosis in vivo and in vitro by increasing the expression of the antiapoptotic protein cFLIP (cellular FLICE inhibitory protein) and by subsequently inhibiting caspase-8 activation. These results suggest that DEX prevents the extensive hepatocellular injury of fulminant hepatic failure induced by cytotoxic cytokines such as TNF-α and anti-Fas antibody.
Results
Pretreatment with DEX prevents primary cultured rat hepatocytes from TNF-α+ActD-induced apoptotic cell death
TNF-α rapidly induces hepatocyte apoptotic cell death in the presence of the transcriptional inhibitor ActD.14 It has been also shown that DEX suppresses spontaneous apoptosis in primary cultured hepatocytes.11 To investigate its antiapoptotic signaling mechanism in TNF-α+ActD-induced hepatocyte apoptosis, we first established the optimal concentration for the inhibition of apoptosis by pretreatment with DEX for 6 h. When incubated with cells in the presence of TNF-α+ActD, DEX pretreatment suppressed apoptotic cell death in a dose-dependent manner as determined by morphologic changes (Figure 1A), DNA fragmentation (Figure 1B), and crystal violet staining (Figure 1C). The maximal inhibition was observed at 5 μM of DEX. As our previous study illustrated,15 the pan-caspase inhibitor Z-VAD-fmk completely suppressed TNF-α+ActD-induced hepatocyte apoptosis (Figure 1C). To examine whether the antiapoptotic effect of DEX is specific to rat hepatocytes or is more general in affecting hepatocyte survival in other species as well, we performed similar experiments using primary cultures of human hepatocytes. Based on cell viability, treatment with increasing concentrations of DEX protected human hepatocytes from TNF-α+ActD-induced apoptosis in a similar fashion to that observed in rat hepatocytes (Figure 1D). Furthermore, we also observed a similar antiapoptotic effect of DEX in primary cultured mouse hepatocytes (data not shown).

DEX prevents rat and human primary hepatocytes from TNF-α+ActD-induced apoptosis. Primary rat hepatocytes were treated with or without 2000 U/ml TNF-α plus 0.2 μg/ml of ActD (TNF-α+ActD) for 12 h following pretreatment with the indicated concentrations of DEX for 6 h. Z-VAD-fmk (100 μM) was cotreated with TNF-α+ActD. (A) Phase-contrast photomicrographs showing cellular morphology of rat hepatocytes; (a) control, (b) TNF-α+ActD, (c) TNF-α+ActD plus 0.1 μM DEX, (d) TNF-α+ActD plus 1 μM DEX, (e) TNF-α+ActD plus 5 μM DEX, and (f) TNF-α+ActD plus 100 μM Z-VAD-fmk. (B) DNA fragmentation was analyzed by agarose gel electrophoresis after genomic DNA was isolated from rat hepatocytes treated with TNF-α+ActD in the presence or absence of DEX or Z-VAD-fmk. (C) Rat hepatocyte viability was measured by crystal violet staining. (D) Primary human hepatocytes were treated with TNF-α+ActD in the presence or absence of the indicated concentrations of DEX. After 12 h, cell viability was measured by crystal violet staining. Data shown in (C) and (D) are the mean±S.D. (n≥3)
DEX pretreatment prevents TNF-α+ActD-induced caspase-3 activation
We next examined the effects of pre- and post-treated DEX on TNF-α+ActD-induced rat hepatocyte apoptosis. Pretreatment with DEX for 6–18 h effectively protected hepatocytes from TNF-α+ActD-induced apoptotic cell death, and this protective effect was gradually decreased by reducing the pretreated time period from 6 to 2 h, whereas no protective effect was observed when DEX was simultaneously treated or post-treated with TNF-α+ActD (Figure 2a). We have previously demonstrated that caspase-3 is activated in hepatocytes challenged with TNF-α+ActD and that caspase activation is required for hepatocyte apoptosis in response to this proapoptotic stimulus.16 To examine whether pretreated DEX suppressed hepatocyte apoptosis by inhibiting activation of the caspase cascade, we evaluated caspase-3 activation and activity in cells exposed to TNF-α+ActD following pretreatment with DEX as determined by immunoblotting analysis and colorimetric assay with Ac-DEVD-pNA. Pretreatment with DEX for 6 h inhibited caspase-3 activation and activity in TNF-α+ActD-treated hepatocytes, and this inhibition was decreased in an inverse time-dependent manner (Figure 2b and c). Moreover, concomitant and post-treatment of cells with DEX resulted in no significant inhibition of caspase-3 activation and activity. One of the established substrates for caspase-3 protease in cells is poly(ADP-ribose) polymerase (PARP), which is cleaved from a 116 kDa intact protein into 85 and 31 kDa fragments during apoptosis.17 The cleaved product (85 kDa) of PARP was detected in the lysate of TNF-α+ActD-treated hepatocytes by Western blot (Figure 2d). PARP cleavage was almost completely inhibited by DEX pretreatment for 6 h, and this inhibition was gradually reversed by decreasing the time period of DEX pretreatment. Together, these findings indicate that DEX pretreatment suppresses TNF-α+ActD-induced apoptosis at a site upstream of caspase-3 activation.

Pretreatment, but not post-treatment, with DEX inhibits TNF-α+ActD-induced apoptosis and caspase-3 activation. Rat hepatocytes were incubated with TNF-α+ActD before or after treatment with 5 μM DEX. (a) After 12 h incubation, cell viability was measured by crystal violet staining. (b) After 6 h incubation, cells were collected and cell lysates were prepared by three cycles of freeze and thaw. Caspase-3 activation was determined by Western blot analysis. (c) Casapase-3-like enzyme (DEVDase) activity was determined using a colorimetric assay with Ac-DEVD-pNA as substrate. (d) PARP cleavage was determined by immunoblotting analysis. Data shown in (a) and (b) are the mean±S.D. (n=3)
DEX pretreatment prevents TNF-α+ActD-induced caspase-8 and -9 activation and cytochrome c release
Since caspase-3 can be activated by proteolytic activation of caspase-9 following the formation of the cytoplasmic apoptosome by the interaction of procaspase-9, cytosolic cytochrome c, Apaf-1, and dATP,18 we examined the effect of DEX pretreatment on caspase-9 activity and mitochondrial cytochrome c release. DEX pretreatment significantly inhibited caspase-9 activity and the redistribution of mitochondrial cytochrome c to the cytosol compared with control (Figure 3a and b), which are closely correlated with its inhibitory effects on caspase-3 activation and activity (Figure 2b and c). Mitochondrial cytochrome c release during apoptosis induced by cytotoxic cytokines including TNF-α is believed to be induced by caspase-8-dependent cleavage of cytosolic p22 Bid into the p15 fragment, which translocates into the mitochondrial membrane and induces cytochrome c release.18 We next tested whether DEX pretreatment inhibits caspase-8 activation/activity and Bid cleavage. As shown in Figure 3c and d, caspase-8 activation/activity and Bid cleavage were markedly increased in cytosolic extracts from hepatocytes treated with TNF-α+ActD, and these biochemical events were significantly inhibited by pretreatment with DEX. These results indicate that pretreatment with DEX interferes with the death signal pathway via the suppression of caspase-8 activation induced by TNF-α+ActD.

Pretreatment with DEX inhibits TNF-α+ActD-induced cytochrome c release, activation of caspase-8, and Bid cleavage. Hepatocytes were treated with TNF-α+ActD following pretreatment with DEX for the indicated time periods. After 6 h, cells were collected and cytosolic fractions and cell lysates were prepared. (a) Caspase-9-like enzyme (LEHDase) activity was measured in the cell lysates by colorimetric assays using the chromogenic substrate 200 μM Ac-LEHD-pNA. (b) Cytochrome c release from mitochondria was determined in the cytosolic fractions by Western blot analysis. (c) Caspase-8 activation and activity (IETDase) were determined in the cell lysates by immunoblotting and colorimetric assays with Ac-IETD-pNA as substrate, respectively. (d) Bid cleavage was determined by Western blot analysis. Data shown for caspase activities are the mean±S.D. (n=3)
DEX does not protect against non-death receptor-mediated hepatocyte apoptosis
Since caspase-8 activation and Bid cleavage are key events in death receptor-mediated apoptosis, we next examined whether DEX could protect hepatocytes from non-death receptor-mediated apoptosis. Hepatocytes were incubated with the proapoptotic drugs cisplatin and staurosporine for 18 h following pretreatment with or without DEX, and cell viability was determined. These proapoptotic drugs effectively induced hepatocyte apoptosis in nonpretreated cells, which is comparable to that induced by TNF-α+ActD, and the apoptotic effects of these drugs were not significantly prevented by DEX pretreatment (Figure 4a). Cisplatin and staurosporine significantly increased DEVDase (caspase-3-like enzyme) activity in control hepatocytes, and this drug-mediated increase in DEVDase activity was not significantly inhibited by DEX pretreatment (Figure 4b). However, these drugs did not increase IETDase (caspase-8-like enzyme) activity, which is an apical signaling mediator in the death receptor-mediated apoptosis pathway, in both control and DEX-pretreated cells (Figure 4c). These results suggest that DEX pretreatment exerts a protective effect on death receptor-mediated apoptosis, but does not significantly inhibit nonreceptor-mediated cell death.

DEX did not protect hepatocytes from apoptosis induced by cisplatin and staurosporine. Hepatocytes were treated with cisplatin (CisPt, 100 μg/ml) or staurosporine (STS, 25 μM) following pretreatment with or without DEX for 6 h. (a) After 18 h, cell viability was measured by crystal violet staining. After 10 h, (b) casapase-3-like enzyme (DEVDase) and (c) caspase-8-like enzyme (IETDase) activities were determined in the cell lysates using a colorimetric assay with Ac-DEVD-pNA and Ac-IETD-pNA as substrates, respectively. Hepatocytes were also incubated with TNF-α+ActD as a positive control. After 12 and 6 h, cell viability and caspase activity were determined, respectively. All data shown are the mean±S.D. (n≥3)
DEX increases cFLIP expression by transcriptional activation
Antiapoptotic Bcl-2 family proteins such as Bcl-2 and Bcl-xL, cIAP, and the activation of the serine/threonine kinase Akt play an important role in inhibiting apoptosis by blocking mitochondrial cytochrome c release and caspase activation.19 We explored whether DEX could suppress apoptosis and apoptotic signaling by increasing the expression of these genes and the activation of Akt. DEX pretreatment did not alter the expression levels of the antiapoptotic genes, Bcl-2, Bcl-xL, Mcl-1, and cIAP as well as endogenous Akt activation via its phosphorylation at Ser473 (Figure 5a). It has been shown that cFLIP, which is structurally similar to procaspase-8, but lacks catalytic activity, acts as a competitive inhibitor of caspase-8 in the DISC formation, which subsequently blocks apical caspase-8 activation.20 We next examined whether DEX regulates cFLIP expression in cultured hepatocytes. DEX pretreatment significantly upregulated cFLIPL-p43 expression in a time-dependent manner, with maximum expression at 6–8 h, and thereafter maintained its protein level until 18 h (Figure 5b). In addition, DEX pretreatment increased cFLIPL-p43 expression in a dose-dependent manner (Figure 5c). The increases in cFLIP protein level can be the result of transcriptional/translational activation or inhibition of protein degradation. To test this possibility, we examined the effect of DEX on the steady-state levels of cFLIP protein and mRNA in the presence or absence of inhibitors of transcription and translation (Figure 5d). The DEX-induced increase in cFLIPL-p43 protein level was significantly reduced by the specific inhibitors of transcription and translation, ActD and cycloheximide, respectively. However, the enhancement of cFLIPL/S mRNA by DEX was abrogated by ActD, but not by cycloheximide, as determined by RT-PCR and quantitative real-time PCR. Thus, these data indicate that DEX upregulates cFLIPL-p43 expression at the transcriptional step.

DEX upregulates cFLIP expression by transcriptional activation. (a) Hepatocytes were treated with various concentrations of DEX for 6 h and cell lysates were prepared. In total, 40 μg of protein was separated on SDS-PAGE. The protein levels of Bcl-xL, Bcl-2, Mcl-1, cIAP, and phospho-Akt were determined by Western blot analyses using antibodies for each protein. Protein loading was determined by stripping and reprobing the same nitrocellulose membrane for the total protein level of Akt or actin. (b and c) Hepatocytes were treated with 5 μM DEX or the indicated concentrations of DEX for the indicated time periods or 6 h. Cells were harvested and cell lysates were prepared. After 40 μg of protein was separated on SDS–PAGE, the protein level of cFLIP was determined by immunoblotting analysis. The same membrane was used for measuring the protein level of actin. (d) Cells were treated with 5 μM DEX in the presence or absence of ActD (0.2 μg/ml) and cycloheximide (CHX, 10 μg/ml) for 6 h. The levels of cFLIP protein and mRNA were determined by Western blot analysis (WB), RT–PCR, and real-time PCR. Data shown are the mean±S.D (n=3)
DEX also suppresses anti-Fas antibody-induced hepatocyte apoptosis by inhibiting DISC formation and caspase-8 activation
The binding of FasL or anti-Fas antibody to the cell surface death receptor Fas rapidly induces a similar apoptotic signaling pathway to that triggered by TNF-α, which is inhibited by the upregulation of cFLIP.20, 21 We examined whether DEX would protect hepatocytes from anti-Fas antibody (Jo2)-induced apoptotic cell death. Incubation of cells with Jo2+ActD increased hepatocyte apoptosis, and this apoptosis was significantly suppressed by pretreatment with DEX (Figure 6a). We next tested the effects of DEX on the activation and activity of caspase-3 and -8. Treatment with Jo2+ActD increased caspase-8 activation and activity, and these increases were significantly inhibited by pretreatment with DEX (Figure 6b and c). DEX pretreatment also inhibited Jo2+ActD-induced increase in caspase-3 activity (Figure 6d), which was correlated with the suppression of upstream caspase-8 activation and activity. Upon triggering by FasL or agonistic antibody, Fas recruits FADD and procaspase-8 and forms the DISC.18 cFLIP competes with procaspase-8 at the level of DISC formation.21 We next investigated whether DEX inhibits the recruitment of procaspase-8 to the DISC. We found that FADD is recruited to the DISC in hepatocytes stimulated with Jo2, but is absent in this complex in nonstimulated cells (Figure 6e). A significant level of procaspase-8 was found in this complex of Jo2-stimulated hepatocytes, and this protein level was reduced by pretreatment with DEX. However, the DISC contained a low level of cFLIPL-p43 in hepatocytes stimulated with Jo2 alone, and this level was increased by DEX pretreatment. These results indicate that DEX pretreatment blocks the DISC formation through the upregulation of cFLIPL-p43 expression and protects hepatocytes from death receptor-induced apoptosis by blocking the caspase-8-dependent downstream signaling pathway.

Pretreatment with DEX protects hepatocytes from Jo2-induced apoptosis by inhibiting DISC formation and caspase-8 activation. Hepatocytes were incubated with 500 ng/ml Jo2 plus 100 ng/ml ActD (Jo2+ActD) following pretreatment with 5 μM DEX for 6 h. (a) After 12 h, cell viability was measured by crystal violet staining. (b) After 6 h incubation, cells were harvested and cell lysates were prepared. Caspase-8 activation was determined by immunoblotting analysis. (c and d) Activities of caspase-8 and -3 were determined in the cell lysates by colorimetric assays using the chromogenic substrates 200 μM Ac-IETD-pNA (caspase-8-like activity) and 200 μM Ac-DEVD-pNA (caspase-3-like activity), respectively. (e) Membrane fractions from cells stimulated with 2 μg/ml biotinylated Jo2 antibody plus 5 μg/ml strepavidin were solublized in buffer B, and DISC was immunoprecipitated with protein A/G-Sepharose via the Jo2 antibody used for stimulation. Control for DISC isolation was obtained by immunoprecipitating Fas from unstimulated cells using 0.2 μg/ml biotinylated Jo2 antibody plus 0.5 μg/ml streptavidin, considering that ∼10% of the anti-Fas antibodies bound to the cells in the stimulating conditions. Immunoprecipitates were separated by SDS-PAGE and blotted with antibodies for Fas, FADD, caspase-8, and cFLIP. Data shown in (a), (c), and (d) are the mean±S.D (n=3). **P<0.01 versus Jo2 alone
siRNA for cFLIP reversed DEX-mediated protection against TNF-α+ActD-induced hepatocyte apoptosis
To test whether an increase in cFLIP expression is a necessary event for DEX-induced protection against TNF-α+ActD-induced hepatocyte apoptosis, an siRNA approach was employed. Transfection with cFLIP siRNA reduced cFLIPL-p43 expression in hepatocytes treated with or without DEX compared with control siRNA-transfected cells, which did not alter the cFLIPL-p43 protein level compared to untransfected cells (Figure 7a). Specific downregulation of cFLIPL-p43 expression by siRNA was found to significantly decrease the protective effect of DEX on TNF-α+ActD-induced apoptosis and further increase the cytotoxic effect of TNF-α in the absence of DEX, while transfection with control siRNA did not affect TNF-α+ActD-induced apoptosis (Figure 7b). Moreover, transfection with cFLIP siRNA increased TNF-α+ActD-induced caspase-8 activation and reversed the inhibitory effects of DEX on caspase-8 activation compared with control siRNA-transfected cells (Figure 7c). A similar effect on caspase-3 activity was observed in cFLIP siRNA-transfected hepatocytes compared with control siRNA-transfected cells (Figure 7d). In contrast, transfection with control siRNA did not alter apoptosis and caspase-8 activation/activity induced by TNF-α+ActD compared with untransfected cells (Figure 7b–d).

Specific knockdown of cFLIP expression by siRNA inhibits the protective effects of DEX on TNF-α+ActD-induced hepatocyte apoptosis. Hepatocytes were transfected with cFLIP or control siRNAs for 12 h and recovered in fresh medium containing 10% FBS for 40 h. Cells were treated with 5 μM DEX for 6 h, followed by treatment with TNF-α+ActD for 6 and 12 h for determining caspase activation/activity and cell viability, respectively. (a) Cell lysates were prepared from hepatocytes treated with 5 μM DEX for 6 h. The protein level of cFLIP was determined by Western blot analysis. (b) After treatment with TNF-α+ActD for 12 h, cell viability was measured by crystal violet staining. (c and d) Caspase-8 activation and activity was determined in the lysates from hepatocytes treated with TNF-α+ActD for 6 h by Western blot analysis and colorimetric assay, respectively. Data shown in (b) and (d) are the mean±S.D. (n=3). *P<0.02 and **P<0.01
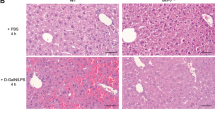
DEX prevents Jo2- and TNF-α+galactosamine (GalN)-induced hepatic injury
Derangement of hepatic apoptosis is considered an important cause for a variety of liver diseases such as viral hepatitis, cholestasis, and ischemic or toxic liver injury. We and others have showed that administration with the agonistic anti-Fas antibody Jo2 and TNF-α+GalN trigger fulminant hepatic failure by inducing in vivo caspase-dependent apoptosis.22, 23 We determined whether DEX prevents the apoptotic signaling pathway and liver injury in animal models. Intraperitoneal injection with DEX significantly increased cFLIPL-p43 expression in liver tissues compared with controls (Figure 8a). Administration of Jo2 and TNF-α+GalN profoundly increased IETDase and DEVDase activities in liver lysates compared with control, and these increases were significantly inhibited by DEX treatment (Figure 8b and c). In line with these observations, the serum levels of the liver function indicators, AST and ALT, were markedly increased in Jo2- and TNF-α+GalN-treated animals, and these increases were suppressed by administration with DEX (Figure 8d). These results indicate that DEX may prevent animals from hepatotoxicity induced by cytotoxic death receptor activation.

DEX administration prevents liver injury induced by Jo2 and TNF-α+GalN. Mice were i.p. administrated with Jo2 (250 μg/kg) or TNF-α (10 μg/kg) plus GalN (700 mg/kg) following i.p. injection with DEX (10 mg/kg). (a) The protein level of cFLIP was determined in whole liver lysates from DEX-treated mice for 6 h. (b and c) After 4- and 8 h-treatments with Jo2 and TNF-α+GalN, respectively, liver tissues were isolated and whole liver lysates were prepared. Activities of caspase-8 and -3 were determined in the liver lysates by colorimetric assays using the chromogenic substrates Ac-IETD-pNA (caspase-8-like activity) and Ac-DEVD-pNA (caspase-3-like activity), respectively. (d) After treatments with Jo2 and TNF-α+GalN for 4 and 12 h, respectively, blood was collected and plasma was prepared by centrifugation at 4000 × g for 30 min. The plasma levels of AST and ALT were determined. Data shown in (b), (c), and (d) are the mean±S.D. (n=6 mice per group). **P<0.01 versus Jo2 or TNF-α+GalN alone
Discussion
This study was undertaken to elucidate the antiapoptotic effect and molecular mechanism of DEX on death receptor-induced hepatocyte apoptosis. Our data demonstrate that DEX inhibits TNF-α-induced in vitro hepatocyte apoptosis and in vivo liver injury by inhibiting the activation of caspase-8, -9, and -3 and mitochondrial cytochrome c release. DEX also inhibited anti-Fas antibody-induced hepatocyte apoptosis by blocking DISC formation and caspase-8 activation. Furthermore, DEX upregulated the expression level of the antiapoptotic protein cFLIPL-p43 in vitro and in vivo. The antiapoptotic effect of DEX was abrogated by specific downregulation of cFLIP expression using siRNA. These results indicate that the upregulation of cFLIP expression may play a key role in the antiapoptotic and protective action of DEX in cell surface death receptor-induced hepatocyte apoptosis.
Glucocorticoids play an important role in normal development and physiological control in animals by modulating the proliferation of various normal and malignant cells.24 The synthetic glucocorticoid DEX has been used in the treatment of lymphoid leukemia and lymphoma and the mechanism underlying this treatment is the induction of apoptosis.25 However, DEX can inhibit apoptosis in some cells such as neutrophils and tumors.6, 12, 26 The antiapoptotic effect by DEX has been shown to be due to the upregulation of the Bcl-2 family proteins such as Bcl-2 and Bcl-xL in hepatocytes.8 These protein levels have been shown to be upregulated in hepatocytes only after long-term treatment with DEX (i.e. 3 days), and this treatment decreased the susceptibility of hepatocytes to spontaneous apoptosis.8 However, we here showed that pretreatment of hepatocytes with DEX for 6–18 h almost fully prevents cells from acute apoptosis (within 18 h) induced by TNF-α and Jo2 in the presence of ActD. In this experimental condition, DEX pretreatment did not alter the protein levels of the antiapoptotic genes such as Bcl-2, Bcl-xL, Mcl-1, and cIAP and the activation of the antiapoptotic serine/threoine kinase Akt, indicating that these genes are not involved in DEX-mediated hepatoprotection. It is possibile that DEX can inhibit caspase-8 activation through the interaction between TNF-α and its cytotoxic receptor TNF-R1. However, we could not find that DEX pretreatment affected the protein level of TNF-R1 in hepatocytes and the cytotoxicity of TNF-α in the culture media of hepatocytes (data not shown). Our results also revealed that DEX pretreatment did not significantly protect hepatocytes from apoptotic cell death induced by cisplatin and staurosporine (Figure 4), which do not essentially require caspase-8 activation for the apoptotic signal pathway. These drugs markedly elevated DEVDase, but not IETDase activity, and this drug-mediated increase in DEVDase activity was not effectively inhibited by DEX pretreatment. However, DEX significantly inhibited the activation/activity of caspase-8 and -3 induced by TNF-α+ActD and Jo2+ActD, indicating that DEX specifically inhibits death receptor-mediated apoptosis. These results suggest that DEX inhibits the activation of caspase-8, which is an apical signal mediator in death receptor-induced apoptosis. cFLIP is known to be a cellular inhibitor for caspase-8 activation in death receptor-induced apoptosis.21 We here showed that DEX pretreatment significantly upregulated the cFLIPL-p43 protein level in a time-dependent manner, which was highly correlated with its antiapoptotic effect. Therefore, our results suggest that DEX can inhibit death receptor-mediated hepatocyte apoptosis via the upregulation of the antiapoptotic gene cFLIP.
Several studies have demonstrated that cFLIP inhibits apoptosis induced by death receptor-activating ligands such as TNF-α, FasL,27 and TRAIL.27, 28, 29 cFLIPL is structurally similar to procaspase-8 because it contains two amino-terminal death effector domains and a carboxyl-terminal caspase homology domain, but it lacks the cysteine within the active site essential for the catalytic activity of caspase-8, resulting in no enzymatic activity.20 Thus, cFLIPL competitively inhibits the recruitment of caspase-8 to the cytosolic death domain of the cytotoxic receptor. Therefore, cFLIPL can be recruited to the Fas- and TNF-α-dependent DISC and inhibits full cleavage and release of active caspase-8 from the DISC.21, 30 The suppression of caspase-8 activation inhibits Bid cleavage and mitochondrial cytochrome c release and subsequently suppresses the formation of an apoptosome formed by the interaction with Apaf-1, procaspase-9, and dATP. Therefore, an increase in cFLIPL expression inhibits caspase-9 activation, resulting in the suppression of downstream signaling cascades including caspase-3 activation. We here showed that DEX pretreatment increased cFLIPL-p43 expression in hepatocytes and inhibited the recruitment of procaspase-8 to the DISC in hepatocytes treated with Jo2, which induces the caspase-8-dependent apoptotic signaling pathway identical to the signal cascade of TNF-α.31 This consequence elicits the protection of hepatocytes from death receptor-induced apoptosis by suppressing caspase-8-activation, Bid cleavage, mitochondrial cytochrome c release, caspase-9 and -3 activation, and PARP cleavage. It has recently been demonstrated that siRNA is a potent mediator of the RNA interference effect, which specifically downregulates target gene expression in animal cells.32 We used this strategy to identify the functional roles of cFLIP in the antiapoptotic effect of DEX on TNF-α-induced hepatocyte apoptosis. Our results indicate that specific knockdown of cFLIPL/S by transfection with cFLIP siRNA effectively inhibited DEX-mediated protection against TNF-α+AcD-induced hepatocyte apoptosis. These results indicate the functional involvement of cFLIPL upregulation in DEX-induced protection from TNF-α+ActD-induced hepatocyte apoptosis.
It has been shown that cFLIP can be upregulated by several cytoprotective factors such as interleukin-427 and lysophospholipids33 through the activation of the PI3K/Akt and NF-κB signaling pathways. Inhibition of PI3K and NF-κB suppressed cFLIP expression and increased apoptosis induced by death receptor-activating cytokines including TNF-α, FasL, and TRAIL. In this study, we showed that DEX did not increase phosphorylation-dependent Akt activation, indicating that the PI3K/Akt pathway is not involved in cFLIP upregulation and antiapoptosis in DEX-treated hepatocytes. We previously showed that DEX inhibits inducible nitric oxide synthase gene expression and nitric oxide production in cultured primary hepatocytes stimulated with TNF-α and interleukin-1β by suppressing NF-κB activation.34, 35 Furthermore, DEX can inhibit the production of proinflammatory cytokines in immune cells by the suppression of NF-κB activation.5 We found that pretreatment of hepatocytes with DEX for 2–12 h inhibited NF-κB activation, as determined by gel-shift assay, in cultured rat primary hepatocytes stimulated with TNF-α (data not shown), suggesting that DEX-mediated upregulation of cFLIP is not due to NF-κB activation. Although not shown here, we also observed that the NF-κB inhibitor pyrrolidine dithiocarbamate did not alter the DEX-induced increase in cFLIPL-p43 expression and protection against TNF-α+ActD-induced hepatocyte apoptosis, indicating no involvement of NF-κB activation in DEX-induced antiapoptosis. These observations indicate that upregulation of cFLIPL-p43 by DEX is not associated with either the PI3K/Akt or NF-κB signaling pathways. Previous studies have shown that DEX can transcriptionally increase the expression levels of several genes in hepatocytes, such as sulfotransferase,36 cytochrome P450 3A1,37 connexin,38 and multidrug-resistance protein 2.39 Similarly, we here showed that increased cFLIP mRNA and protein levels by DEX treatment were markedly suppressed by the transcription inhibitor ActD, indicating that DEX increased cFLIP expression at the transcriptional step. The molecular mechanism for DEX-dependent hepatic cFLIP expression is currently under investigation.
The extensive hepatocellular injury of fulminant hepatic failure is thought to be mediated, in part, by ligation of death receptors with their ligands including TNF-α40 and FasL.41 TNF-α and Jo2 (anti-Fas antibody) induce hepatic apoptosis, which is considered an important cause for a variety of liver diseases such as viral hepatitis, cholestasis, and ischemic or septic liver injury.1, 42 Antiapoptotic drugs or genes including caspase inhibitors have been suggested to prevent fulminant hepatic failure by inhibition of hepatic apoptosis.43 Our data showed that DEX administration in vivo upregulated cFLIPL expression and inhibited Jo2- and TNF-α+GalN-induced increases in caspase-8 and -3 activities, indicating that DEX can block hepatocellular apoptotic cell death in the some clinical settings such as endotoxin-mediated liver failure. This study was focused on determining if administration of DEX into animals can ameliorate liver damage following exposure to Jo2 and TNF-α where liver failure is more gradual in onset. We found that DEX inhibited Jo2- and TNF-α+GalN-mediated increases in plasma AST and ALT, which are indicators of liver function. These results suggest that DEX protects unwanted hepatic cell death in pathological conditions by suppression of caspase-8 activation through the upregulation of cFLIP.
In conclusion, we demonstrated that a possible molecular mechanism for the antiapoptotic effect of DEX is likely the suppression of death receptor-induced DISC formation, caspase-8 activation, and mitochondrial cytochrome c release by increased levels of cFLIPL. Our results suggest that DEX inhibits the extensive hepatocellular injury of fulminant hepatic failure induced by cytotoxic cytokines such as TNF-α and FasL, thereby enabling future clinical therapies such as liver transplantation and septic liver failure.
Materials and Methods
Materials
William's medium E, penicillin, streptomycin, L-glutamate, and HEPES were purchased from Invitrogen (Grand Island, NY, USA). Insulin was obtained from Lilly Inc. (Indianapolis IN, USA) and calf serum was purchased from Hyclone Laboratories (Logan, UT, USA). Cytochrome c antibody and caspase-8 antibody were obtained from PharMingen (San Diego, CA, USA). Polyclonal caspase-3 antibody, PARP antibody, and cFLIPL/S antibody (H-202) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody for phospho-specific Akt (Ser-473) was obtained from Transduction Laboratories (Lexington, KY, USA). Z-VAD-fmk, Ac-DEVD-pNA, Ac-LEHD-pNA, and Ac-IETD-pNA were obtained from Alexis Corporation (San Diego, CA, USA). Rabbit polyclonal anti-Bid antibody was prepared using full-length mouse Bid.44 All other reagents were purchased from Sigma, unless indicated otherwise.
Preparation of primary hepatocytes and cell culture
Rat hepatocytes were isolated by collagenase perfusion from male Sprague–Dawley rats.15 In accordance with institutional review guidelines of the University of Pittsburgh, human hepatocytes were obtained by collagenase digestion of human donor livers not used for transplantation.45 Highly purified hepatocytes (>98% purity and >98% viability by trypan blue exclusion) were suspended in Williams medium E supplemented with 10% calf serum, 1 μM insulin, 2 mM L-glutamine, 15 mM HEPES (pH 7.4), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were plated in 100 mm Petri dishes (5 ml/dish) and 12-well plates (1 ml/well) at a concentration of 1 × 106 and 2 × 105 cells/ml, respectively, and cultured in a CO2 incubator (5% CO2/95% air) at 37°C for 16 h. Cells were pretreated with or without DEX and incubated with 2000 U/ml TNF-α plus 0.2 μg/ml actinomycin D (ActD) (TNF-α+ActD) or 0.5 μg/ml anti-Fas antibody (Jo2) plus 0.2 μg/ml ActD.
Assay for cell viability and DNA fragmentation
Cell viability was determined by crystal violet staining.15 In brief, cells were stained with 0.5% crystal violet in 30% ethanol and 3% formaldehyde for 10 min at room temperature. Plates were then washed four times with tap water. Cells were lysed with 1% SDS solution, and dye uptake was measured at 550 nm using a microplate reader. To assess apoptosis, isolation and electrophoresis of genomic DNA were performed as described previously.15
Enzyme activity assay
Caspase activity was evaluated by measuring proteolytic cleavage of chromogenic substrate as described previously.15 Ac-DEVD-pNA, Ac-LEHD-pNA, and Ac-IETD-pNA were used as the substrates for DEVDase, LEHDase (caspase-9-like enzyme), and IETDase, respectively. Briefly, cell lysate (100 μg of protein) was added into Tris-HCl buffer (pH7.4) containing 200 μM Ac-DEVD-pNA, 200 μM Ac-LEHD-pNA, or 200 μM Ac-IETD-pNA in a final volume of 100 μl. The reaction mixture was incubated at 37°C for 1 h. The increase in absorbance of enzymatically released pNA was measured at 405 nm in a microplate reader.
Western blot analysis
Cells were harvested, washed twice with ice-cold PBS, and resuspended in 20 mM Tris-HCl buffer (pH 7.4) containing a protease inhibitor mixture (0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml pepstatin A, and 1 μg/ml chymostatin). Cytosolic fractions were prepared for cytochrome c release by homogenization in an isotonic sucrose solution using a Dounce homogenizer and differential centrifugation, as described previously.46 Whole-cell lysates were prepared for immunoblotting analysis of caspases and PARP by sonication and centrifugation at 12 000 × g for 20 min at 4°C. Protein concentration was determined with the BCA assay (Pierce) with bovine serum albumin as standard. Proteins (40 μg) were separated on SDS-PAGE and then transferred onto nitrocellulose. The membranes were hybridized with antibodies against cytochrome c, caspase-3 and -8, Bcl-2, Bcl-xL, Mcl-1, cIAP, cFLIPL/S, p-Akt, Akt, and actin, and protein bands were visualized by exposing to X-ray film, as described previously.15
DISC isolation
DISC was isolated as described previously.47 In brief, hepatocytes were stimulated with 2 μg/ml biotinylated Jo2 anti-Fas-antibody plus 5 μg/ml strepavidin (Pierce) for 30 min at 37°C. Cells were gently sonicated (5 s bursts, 5 W; Vibracell, Bioblock Scientific in ice-cold buffer A containing 25 mM HEPES, 150 mM NaCl, 1 mM EGTA, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 2 μg/ml chymostatin, and 5 μg/ml α2 macroglobin). After nuclear fraction was removed by centrifugation at 800 × g for 10 min at 4°C, the membrane fraction was obtained from the supernatant by centrifugation at 100 000 × g for 1 h at 4°C. The pellet was solubilized in buffer A containing 1% Triton X-100 and 10% glycerol (buffer B) at 4°C. The solubilizate was then subjected to immunoprecipitation via Jo2 anti-Fas antibody used for stimulation with protein A/G-Sepharose beads (Amersham Pharmacia) at 4°C for 2 h to isolate DISC. Control for DISC isolation was obtained by immunoprecipitating Fas from unstimulated cells using 2 μg/ml Jo2 antibody plus 5 μg/ml streptavidin, considering that ∼10% of the anti-Fas antibodies bound to the cells in the stimulating conditions. The immunoprecipitates were washed four times before eluted from the beads by heating in SDS-PAGE sample buffer at 95°C for 5 min.
Transfection with siRNA
The sequences of mouse cFLIP siRNA and control scrambled siRNA were 5′-AAGTAAAGAGCCAAGATTTGT-3′ (627–647 nt from start codon), which targets both cFLIPL and cFLIPS, and 5′-AACAATCGAGTGTATAGATAG-3′, respectively. Cells were transfected with the double-stranded siRNAs (40 nmol/ml) for 12 h by lipofectamine method according to the manufacturer's protocol (Invitrogen) and recovered in fresh media containing 10% FBS for 40 h. The cells were pretreated with DEX for 6 h and then treated with TNF-α+ActD. The protein levels of cFLIP were analyzed by Western blot analysis, and cell viability was determined by crystal violet staining.
Reverse transcriptase-polymerase chain reaction (RT-PCR) and quantitative real-time PCR analyses
Total RNA was obtained from hepatocytes using a Trizol reagent kit (Invitrogen). In total, 3 μl of the cDNA mixture was used for PCR in 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 0.2 mM dNTPs, 2.5 U of Taq DNA polymerase, and 0.01 nM of primers for cFLIP and β-actin. The amplification was performed in a DNA thermal cycler under the following conditions: denaturation at 94°C for 5 min for the first cycle and for 30 s starting from the second cycle, annealing at 62°C for 30 s, and extension at 72°C for 30 s for 30 cycles. Final extension was performed at 72°C for 10 min. The PCR products were electrophoresed on a 1.5% agarose gel and stained with ethidium bromide. The primers used were 5′-ATCTGGTGATTGAATTGGAG-3′ (sense; 86–111 nt from start codon) and 5′-ATATGATAGCCCAGGGAAGT-3′ (antisense; 495–521 nt from start codon) for cFLIPL/S and 5-TCCTTCGTTGCCGGTCCACA-3 (sense) and 5-CGTCTCCGGAGTCCATCACA-3 (antisense) for β-actin. The mRNA expression level was quantified by real-time PCR using the SYBR Green PCR Master Mix in an ABI PRISM 7700 Sequence Detector (Applied Biosystems) with 10 μl of diluted (1 : 25) cDNA and gene-specific primers according to the manufacturer's instructions. cFLIP mRNA expression levels were quantified using the threshold cycle method. Values were then normalized to the relative amounts of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase.
Animal experiment
All procedures performed on animals were in accordance with the guidelines of the University Animal Care and Use Committee. BALB/c mice (6–8 weeks, 20 g) were intraperitoneally (i.p.) injected with 10 mg/kg DEX. At 6 h following DEX pretreatmenr, mice were i.p. injected with Jo2 (250 μg/kg) or TNF-α (10 μg/kg)+GalN (700 mg/kg). After 4- and 8 h-treatments with Jo2 and TNF-α+GalN, respectively, animals were placed under inhaled isoflurane anesthesia for isolation of blood and liver tissue. The latter was snap-frozen and stored at −80°C until it was assessed for caspase activity. Plasma was prepared from whole blood by centrifugation at 4000 × g for 30 min at 4°C. Plasma levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using enzyme assay kits.
Data analysis and statistics
The data are presented as means±S.D. from more than three independent experiments. Statistical comparisons between groups were performed using the Student's t-test.
Abbreviations
- DEX:
-
dexamethasone
- TNF-α:
-
tumor necrosis factor-alpha
- ActD:
-
actinomycin
- GalN:
-
galactosamine
- cFLIP:
-
cellular FLICE inhibitory protein
- TNF-R:
-
TNF-α receptor
- FADD:
-
Fas-associated death domain-containing protein
- DISC:
-
death-inducing signaling complex
- IETDase:
-
caspase-8-like enzyme
- LEHDase:
-
caspase-9-like enzyme
- DEVDase:
-
caspase-3-like enzyme
- CAD:
-
caspase-activated DNase
- PARP:
-
poly(ADP-ribose) polymerase
References
Luedde T, Liedtke C, Manns MP and Tautwein C (2002) Losing balance: cytokine signaling and cell death in the context of hepatocyte injury and hepatic failure. Eur. Cytokine Network 4: 377–383
Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ and Brenner DA (2004) Differential requirement for c-Jun NH2-terminal kinase in TNF-α- and Fas-mediated apoptosis in hepatocytes. FASEB J. 18: 720–722
Wajant H, Pfizenmaier K and Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ. 10: 45–65
Sakahira H, Enari M and Nagata S (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391: 96–99
Baeuerle PA and Baichwal VR (1997) NF-κB as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 65: 111–137
Kato T, Takeda Y, Nakada T and Sendo F (1995) Inhibition by dexamethasone of human neutrophil apoptosis in vitro. Nat. Immun. 14: 198–208
Yamamoto M, Fukuda K, Miura N, Suzuki R, Kido T and Komatsu Y (1998) Inhibition by dexamethasone of transforming growth factor β1-induced apoptosis in rat hepatoma cells: a possible association with Bcl-xL induction. Hepatology 27: 959–966
Bailly-Maitre B, de Sousa G, Boulukos K, Gugenheim J and Rahmani R (2001) Dexamethasone inhibits spontaneous apoptosis in primary cultures of human and rat hepatocytes via Bcl-2 and Bcl-xL induction. Cell Death Differ. 8: 279–288
Cifone MG, Migliorati G, Parroni R, Marchetti C, Millimaggi D, Santoni A and Riccardi C (1999) Dexamethasone-induced thymocyte apoptosis: apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood 93: 2282–2296
Zacharchuk CM, Mercep M, Chakraborti PK, Simons Jr SS and Ashwell JD (1990) Programmed T lymphocyte death. Cell activation- and steroid-induced pathways are mutually antagonistic. J. Immunol. 145: 4037–4045
Bailly-Maitre B, de Sousa G, Zucchini N, Gugenheim J, Boulukos KE and Rahmani R (2002) Spontaneous apoptosis in primary cultures of human and rat hepatocytes: molecular mechanisms and regulation by dexamethasone. Cell Death Differ. 9: 945–955
Suffys P, Beyaert R, De Valck D, Vanhaesebroeck B, Van Roy F and Fiers W (1991) Tumour-necrosis-factor-mediated cytotoxicity is correlated with phospholipase-A2 activity, but not with arachidonic acid release per se. Eur. J. Biochem. 195: 465–475
Wu YL, Jiang XR, Lillington DM, Newland AC and Kelsey SM (2000) Upregulation of lipocortin 1 inhibits tumour necrosis factor-induced apoptosis in human leukaemic cells: a possible mechanism of resistance to immune surveillance. Br. J. Haematol. 111: 807–816
Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G and Wendel A (1994) Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-α requires transcriptional arrest. J. Immunol. 153: 1778–1788
Kim YM, Talanian RV and Billiar TR (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272: 31138–31148
Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM and Billiar TR (2000) Nitric oxide prevents tumor necrosis factor α-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology 32: 770–778
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin TT, Yu VL and Miller DK (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43
Budihardjo I, Oliver H, Lutter M, Luo X and Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15: 269–290
Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K, Bharti A, Yuan ZM, Saxena S, Weichselbaum R, Nalin C and Kufe D (1997) Role for Bcl-xL as an inhibitor of cytosolic cytochrome c accumulation in DNA damage-induced apoptosis. Proc. Natl. Acad. Sci. USA 94: 6939–6942
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE and Tschopp J (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388: 190–195
Krueger A, Schmitz I, Baumann S, Krammer PH and Kirchhoff S (2001) Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 276: 20633–20640
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA and Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891
Saavedra JE, Billiar TR, Williams DL, Kim YM, Watkins SC and Keefer LK (1997) Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-α-induced apoptosis and toxicity in the liver. J. Med. Chem. 40: 1947–1954
Tsai MJ and O’Malley BW (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu. Rev. Biochem. 63: 451–486
Harmon JM, Norman MR, Fowlkes BJ and Thompson EB (1979) Dexamethasone induces irreversible G1 arrest and death of a human lymphoid cell line. J. Cell Physiol. 98: 267–278
Chaya D, Fougere-Deschatrette C and Weiss MC (1997) Liver-enriched transcription factors uncoupled from expression of hepatic functions in hepatoma cell lines. Mol. Cell. Biol. 17: 6311–6320
Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G, Messina A, Peschle C and De Maria R (2004) IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of anti-apoptotic proteins. J. Immunol. 172: 5467–5477
Leverkus M, Neumann M, Mengling T, Rauch CT, Brocker EB, Krammer PH and Walczak H (2000) Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 60: 553–559
Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K and Nakano T (2003) Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab. Invest. 83: 1033–1043
Thome M and Tschopp J (2001) Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1: 50–58
Baker SJ and Reddy EP (1996) Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene 12: 1–9
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K and Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498
Kang YC, Kim KM, Lee KS, Namkoong S, Lee SJ, Han JA, Jeoung D, Ha KS, Kwon YG and Kim YM (2004) Serum bioactive lysophospholipids prevent TRAIL-induced apoptosis via PI3K/Akt-dependent cFLIP expression and Bad phosphorylation. Cell Death Differ. 11: 1287–1298
Geller DA, Nussler AK, Di Silvio M, Lowenstein CJ, Shapiro RA, Wang SC, Simmons RL and Billiar TR (1993) Cytokines, endotoxin, and glucocorticoids regulate the expression of inducible nitric oxide synthase in hepatocytes. Proc. Natl. Acad. Sci. USA 90: 522–526
De Vera ME, Taylor BS, Wang Q, Shapiro RA, Billiar TR and Geller DA (1997) Dexamethasone suppresses iNOS gene expression by upregulating IκBα and inhibiting NF-κB. Am. J. Physiol. 273: G1290–G1296
Duanmu Z, Locke D, Smigelski J, Wu W, Dahn MS, Falany CN, Kocarek TA and Runge-Morris M (2002) Effects of dexamethasone on aryl (SULT1A1)- and hydroxysteroid (SULT2A1)-sulfotransferase gene expression in primary cultured human hepatocytes. Drug Metab. Dispos. 30: 997–1004
Burger HJ, Schuetz JD, Schuetz EG and Guzelian PS (1992) Paradoxical transcriptional activation of rat liver cytochrome P-450 3A1 by dexamethasone and the antiglucocorticoid pregnenolone 16α-carbonitrile: analysis by transient transfection into primary monolayer cultures of adult rat hepatocytes. Proc. Natl. Acad. Sci. USA 89: 2145–2149
Ren P, de Feijter AW, Paul DL and Ruch RJ (1994) Enhancement of liver cell gap junction protein expression by glucocorticoids. Carcinogenesis 15: 1807–1813
Kubitz R, Warskulat U, Schmitt M and Haussinger D (1999) Dexamethasone- and osmolarity-dependent expression of the multidrug-resistance protein 2 in cultured rat hepatocytes. Biochem. J. 340 (Part 3): 585–591
Muto Y, Nouri-Aria KT, Meager A, Alexander GJ, Eddleston AL and Williams R (1988) Enhanced tumour necrosis factor and interleukin-1 in fulminant hepatic failure. Lancet 2: 72–74
Kondo T, Suda T, Fukuyama H, Adachi M and Nagata S (1997) Essential roles of the Fas ligand in the development of hepatitis. Nat. Med. 3: 409–413
Yoneda M, Wada K, Katayama K, Nakajima N, Iwasaki T, Osawa E, Mukasa K, Yamada Y, Blumberg RS, Sekihara H and Nakajima A (2004) A novel therapy for acute hepatitis utilizing dehydroepiandrosterone in the murine model of hepatitis. Biochem. Pharmacol. 68: 2283–2289
Kim KM, Kim YM, Park M, Park K, Chang HK, Park TK, Chung HH and Kang CY (2000) A broad-spectrum caspase inhibitor blocks concanavalin A-induced hepatitis in mice. Clin. Immunol. 97: 221–233
Wang K, Yin XM, Chao DT, Milliman CL and Korsmeyer SJ (1996) BID: a novel BH3 domain-only death agonist. Genes Dev. 10: 2859–2869
Nussler AK, Di Silvio M, Billiar TR, Hoffman RA, Geller DA, Selby R, Madariaga J and Simmons RL (1992) Stimulation of the nitric oxide synthase pathway in human hepatocytes by cytokines and endotoxin. J. Exp. Med. 176: 261–264
Kim YM, Chung HT, Simmons RL and Billiar TR (2000) Cellular non-heme iron content is a determinant of nitric oxide-mediated apoptosis, necrosis, and caspase inhibition. J. Biol. Chem. 275: 10954–10961
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH and Peter ME (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14: 5579–5588
Acknowledgements
This work was supported by Vascular System Research Center grant from Korea Science and Engineering Foundation.
Author information
Authors and Affiliations
Author notes
H-Y Oh, S Namkoong: These authors contributed equally to this work.
Corresponding author
Additional information
Edited by S Kaufmann
Rights and permissions
About this article
Cite this article
Oh, HY., Namkoong, S., Lee, SJ. et al. Dexamethasone protects primary cultured hepatocytes from death receptor-mediated apoptosis by upregulation of cFLIP. Cell Death Differ 13, 512–523 (2006). https://doi.org/10.1038/sj.cdd.4401771
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401771
Keywords
This article is cited by
-
A characterization of cortisol level and adrenal reservation in human cardiopulmonary arrest: systematic review and meta-analysis
Systematic Reviews (2021)
-
Galactose protects hepatocytes against TNF-α-induced apoptosis by promoting activation of the NF-κB signaling pathway in acute liver failure
Laboratory Investigation (2015)
-
Glucocorticoids protect renal mesangial cells from apoptosis by increasing cellular sphingosine-1-phosphate
Kidney International (2010)
-
Involvement of the ABC-transporter ABCC1 and the sphingosine 1-phosphate receptor subtype S1P3 in the cytoprotection of human fibroblasts by the glucocorticoid dexamethasone
Journal of Molecular Medicine (2009)
-
Glucocorticoids inhibit the apoptotic actions of UV-C but not Fas ligand in hepatoma cells: direct evidence for a critical role of Bcl-xL
Cell Death & Differentiation (2007)
