
Abstract
Promyelocytic leukaemia (PML) nuclear bodies (NBs) are macromolecular nuclear domains present in virtually every mammalian cell. PML nuclear bodies (PML-NBs) were functionally linked to various fundamental cellular processes, including transcriptional control, tumour suppression and apoptosis regulation. Supporting the important function of PML and its associated NBs in apoptosis regulation, several apoptotic regulators localise to PML-NBs, and cells from PML-deficient mice show severe apoptotic defects, including induction of genotoxic stress and death receptor CD95 (Fas/APO-1) activation. Based on the current literature, we hypothesise that PML-NBs regulate apoptosis through different molecular mechanisms, on the one hand by acting as macromolecular scaffolds for recruitment and post-translational modification of the apoptotic key regulator p53, and on the other by regulating the subcellular bioavailability and quality of some apoptotic signal transducers.
Similar content being viewed by others


Introduction
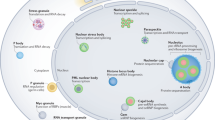
The mammalian cell nucleus harbours a number of subnuclear compartments among the family of nuclear bodies (NBs).1 One particular class of NBs are the promyelocytic leukaemia nuclear bodies (PML-NBs, also termed PML oncogenic domains, PODs, Kremer bodies, nuclear domain 10, ND10, NBs or nuclear dots, NDs). PML-NBs are dynamic macromolecular structures altering their number, size and content in response to diverse stimuli such as viral infections, extracellular signals and genotoxic stress.2 Under normal growth conditions, almost every mammalian cell usually harbours between 10 and 30 doughnut-like-shaped PML-NBs with a diameter ranging between 0.2 and 1 μm.3,4,5,6,7 Besides a number of viral proteins, to date more than 40 different cellular proteins have been found in association with PML-NBs (Figure 1). Although the molecular function of PML-NBs is currently not clear, there is accumulating evidence that PML-NBs are regulatory domains involved in various biological processes, including protein degradation,5 transcriptional regulation,8,9 cell growth,10,11 antiviral response,12,13 cellular senescence,14,15,16,17 tumour suppression,18 DNA repair19,20 and apoptosis.21,22,23,24,25 However, how can one explain that so many different and unrelated processes are linked to these nuclear domains? Since PML-NBs contain several dozens of functionally different residents (Figure 1), the current model is that PML-NBs play an important role in the regulation of these processes by controlling the function of its residents by altering their post-translational modification pattern and regulating their subcellular distribution.

Cellular PML-NB components. A list of cellular proteins identified to localise at PML-NBs. Currently, the PML and Sp100 proteins are the only known constitutive PML-NB residents. All other indicated components either transiently localise to PML-NBs or are recruited to PML-NBs under specific conditions, including treatment with type I and II interferons, genotoxic stress or oncogenic transformation and induction of premature cellular senescence. The various PML-NB components have functions in transcriptional regulation, maintenance of genomic stability, post-translational protein modification, protein degradation, DNA repair and apoptosis
PML and its associated NBs
PML was originally identified in leukaemic blasts from patients suffering from acute promyelocytic leukaemia (APL). In APL blasts, PML-NBs are delocalised into multiple tiny microspeckeled NBs,26 in most cases due to the expression of the oncogenic PML-RARα fusion protein, which is the product of a reciprocal chromosomal translocation.27,28,29,30 PML-RARα causes APL through a multifunctional mechanism involving RARα target gene repression,31 inhibition of apoptosis32 and epigenetic gene silencing.33 Treatment of APL patients with all-trans retinoic acid (ATRA) or the chemotherapeutic drug arsenic trioxide (As2O3) leads to PML-RARα degradation, reformation of PML-NBs and terminal differentiation to myelocytes, or in case of As2O3 treatment to apoptosis of the APL blasts, which is followed by disease remission.26,32
The tumour suppressor protein PML belongs to the family of RING domain containing proteins34,35 and is expressed in at least seven different isoforms, which differ in their function and subcellular localisation.36 Unravelling the functional differences of the PML isoforms is currently an intensively investigated topic in PML research. Cells from PML-deficient mice lack PML-NBs and the PML-NB components Daxx, small ubiquitin-like modifier-1 (SUMO-1) and CREB-binding protein (CBP) are mislocalised throughout the nucleoplasm. The exogenous expression of PML relocalises these factors to PML-NBs,5,37,38 indicating that PML, although it appears not to bind all its PML-NB residents, establishes a supramolecular scaffold necessary for recruiting diverse proteins to this nuclear domain.
Recently, it has been reported that the PML-NB component eIF4E localises to NBs in PML−/− cells, and exogenously expressed PML distributes to eIF4E bodies in PML−/− cells, demonstrating that eIF4E forms NBs independent of PML.2,39 This finding suggests that eIF4E bodies might represent the underlying structure where PML-NBs are assembled. However, more detailed investigations using, for example, nontransformed primary human diploid cells are needed to generalise this interesting finding.
Role of SUMO in PML-NB formation and transcriptional regulation
The assembly of PML into macromolecular PML-NBs depends on its covalent post-translational modification with SUMO-1.37,38 SUMO-1 (also termed sentrin, GMP1, SMT3C or ULP1) is a small polypeptide that is covalently attached to PML and several other PML-NB components, including Sp100,40 Daxx,41 p5342,43 and CBP,44 through an enzymatic machinery similar to that of ubiquitin modification.45 SUMOlation requires an activating E1 SUMO-1 activating enzyme (SAE)1/SAE2 heterodimer,46 the conjugating E2 enzyme Ubc947,48 and the recently identified E3 SUMO ligases.49,50,51,52 In contrast to ubiquitination, SUMOlation usually does not lead to protein degradation but regulates protein–protein interactions, subcellular localisation and activity. Interestingly, SUMO-conjugated PML interacts with Daxx and recruits Daxx from chromatin into the PML-NBs and releases the Daxx repressor function.8,53 This mechanism allows the regulation of Daxx function via PML SUMO conjugation and deconjugation. Recently, different SUMO peptidases have been identified in mammals, which differ in their subcellular localisation and target protein specificity. Interestingly, a recently identified SUMO peptidase that deconjugates SUMO from PML has been shown to regulate PML-NB assembly and transcription.54 Similarly, p300/CBP and Sp3 are both regulated in their cofactor function – whether they act as a coactivator or corepressor – via timely controlled SUMO conjugation and deconjugation.44,55 Thus, SUMO is an important factor to control PML-NB assembly and transcription. For a detailed review about the function of SUMO, we refer to some recently published reviews on this topic.45,56,57
Several reports indicate an important function of PML and its NB residents in apoptosis regulation. As discussed below, we hypothesise that PML-NBs regulate apoptosis through different mechanisms, either by serving as transcriptional modification and activation platforms for the tumour suppressor p53, and in addition, by regulating the quality and subcellular bioavailability of apoptotic regulators. As a basis for our discussion of the mechanisms of how PML-NB components participate in apoptosis regulation, we will first briefly outline the different apoptosis pathways.
Apoptosis pathways
Apoptosis is a tightly regulated mechanism to eliminate unwanted or potentially dangerous cells in multicellular organisms during development, tissue homeostasis and immune response.58,59 The physiological meaning of apoptosis is illustrated by a number of human diseases, including cancer, autoimmune disorders and neurodegenerative diseases, which are associated with apoptosis dysregulation.60,61,62,63
Two principle apoptotic pathways exist in mammalian cells, the intrinsic and extrinsic pathways. Both pathways converge on the level of caspase activation. Caspases, aspartate-specific proteases, are the main arm of the apoptotic machinery,64 although caspase-independent apoptosis pathways have also been described.65,66 Caspases are synthesised as procaspases, catalytically inactive precursors (zymogens), and are proteolytically activated either by other caspases and proteases or by a local increase in caspase concentration.67 The intrinsic pathway is regulated by the mitochondria and is activated by translocation of proapoptotic members of the Bcl-2 protein family (such as Bax or truncated Bid) into the mitochondrial membrane,68 which triggers the release of cytochrome c into the cytosol. Cytochrom c binds the apoptosis-activating factor 1 (APAF-1), an evolutionarily conserved proapoptotic factor, in the cytosol in an energy-dependent manner. APAF-1 then assembles into a multimeric caspase activation platform termed ‘apoptosome’, which recruits and activates caspase-9 and thus allows the activation of the downstream effector caspase-3.69
Extrinsic apoptosis pathways are elicited by a large number of cell surface receptors, which are capable of inducing apoptosis upon their multimerisation and activation through their respective ligands or agonistic antibodies.61 The best understood death receptor systems are the tumour necrosis factor receptor (TNF-R)1 and CD95 (Fas/APO-1), members of the death receptor superfamily characterised by an intracellular death domain (DD). Upon their activation, they transmit an apoptotic signal via intracellular recruitment and assembly of a death-inducing signalling complex (DISC).70,71 The CD95 and TNF-R1 DISCs are multiprotein complexes that consist of the adaptor molecules FADD and TNF-R1-associated death domain protein (TRADD) (only in the TNF-R1 receptor system), which in turn mediate recruitment of the death-effector domain (DED) containing initiator procaspases caspase-8 and -10 (only in humans) to the DISC. The procaspases are then autocatalytically cleaved and processed to active caspases,67,72 which activate a caspase cascade finally resulting in proteolysis of various death substrate proteins and the execution of cell death.73
In addition to receptor-induced pathways, nuclear apoptosis pathways also exist. For example, several transcription factors, including p53 or forkhead transcription factors74,75 can induce the expression of proapoptotic target genes that either directly trigger apoptosis via the intrinsic, mitochondrial pathway (e.g. by upregulating proapoptotic Bcl-2 family members76,77,78) or by transcriptional upregulation of death receptors such as CD9579 or p53RDL1,80 which results in extrinsic receptor-mediated apoptosis pathways. Moreover, apoptosis sensitivity can be altered by repressing the target gene expression of antiapoptotic transcription factors, such as NF-κB, which leads to an increased sensitivity towards TNF-α-induced apoptosis.81,82,83
PML-NB components in receptor-mediated apoptosis pathways
PML
Cells derived from PML−/− mice show, in addition to defects in ceramide-induced, γ-irradiation-induced and type I and type II interferon-induced apoptosis, a strongly decreased sensitivity against death receptor-mediated apoptosis through stimulation with CD95L and TNF-α.21 Moreover, injection of CD95-activating antibodies in PML−/− mice resulted in a markedly decreased apoptosis-derived liver failure and increased survival time. Despite the defects in multiple apoptosis pathways, PML−/− mice are viable and show no obvious abnormalities.21 This might reflect the particular importance of PML in apoptosis pathways that are not essential for normal development, reminiscent of the phenotype of the tumour suppressor p53, which also plays a pivotal role only in stress-induced apoptosis pathways.84
Although a role of PML in death receptor-induced apoptosis is likely, there is currently no evidence that PML exerts a direct function in death receptor signalling by participating in receptor proximal events. Thus, one might assume that PML-NBs regulate the bioavailability of signal transduction components participating in death receptor-induced apoptosis pathways either by regulating their expression and/or by controlling their subcellular distribution. In agreement with this assumption, it was recently reported that APL cells respond to ATRA treatment by upregulation of the death ligand tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) leading to paracrine apoptosis of the APL blasts, indicating that PML-RARα suppresses the normal regulation of TRAIL in these cells.85
The CD95 and TNF-R1 can signal cell death via caspase-dependent and -independent pathways.61,65,66 Although the DISC components and caspase-1, -3 and -8 are properly expressed, caspase activation is markedly inhibited upon CD95L or TNF-α stimulation in PML-deficient cells.21 The underlying molecular defect explaining this interesting phenotype is presently unknown. Currently available evidence does not fully exclude the fact that nucleoplasmic or cytoplasmic PML can also directly regulate the apoptotic machinery NB-independently. Taking the defects in PML-deficient cells in various apoptosis pathways into account, this either might be explained by a defect in a general downstream death signalling component, or by various pathway-specific upstream defects. The detailed role of PML in CD95-mediated apoptosis and other death receptor pathways is far from being understood and awaits further investigations. PML also regulates a caspase-independent apoptosis pathway.86 Using an inducible overexpression system, it has been shown that PML can trigger a death pathway independent from de novo protein synthesis that is not sensitive to synthetic caspase inhibitors.86 However, this finding remains to be confirmed under more physiological conditions. An overview of currently known PML-NB-associated proteins and their function in cell surface receptor-mediated or nuclear apoptosis pathways are given in Table 1 and in Figure 2.

Model of PML-NB components and their function in nuclear- and surface receptor-mediated apoptosis pathways. Fractions of the proteins Daxx, TRADD and HIPK3 were shown to localise both into PML-NBs and to the cytoplasmic compartment. Dependent on their subcellular localisation and their molecular context Daxx, HIPK3 and TRADD can either function directly in cell surface receptor-mediated apoptosis pathways70,88,95,100,101,113 or in nuclear apoptosis pathways87,102,105,148 by regulating the target gene expression of proapoptotic transcription factors such as p53 or by suppressing the activity of the antiapoptotic transcription factor NF-B. The regulated PML-NBs recruitment and the release of these proteins may control their bioavailability and might modulate cellular sensitivity towards nuclear or receptor-mediated apoptosis pathways. PM, plasma membrane. For a detailed description, see text
TRADD
Recently, the TNF-R1-associated DD protein TRADD was found to shuttle between the cytoplasm and the nucleus.87 In the cytoplasm, TRADD serves as an adaptor molecule for the recruitment of FADD and caspase-8 to the TNF-R1 DISC.70 In the nuclear compartment, TRADD is found in association with PML-NBs.87 The isolated DD of TRADD, which also mediates the interaction with the TNF-R1 DD,70,88 is responsible for PML-NB localisation and is sufficient to initiate a caspase-independent form of cell death. Apoptosis through TRADD-DD overexpression is in part inhibited by Bcl-XL, a mitochondria-localised antiapoptotic member of the Bcl-2 protein family, and relies on p53 and PML expression.87 These results imply that a pathway targeting the mitochondria is involved in this death mechanism. The physiological relevance and the exact molecular mechanism of TRADD-DD-induced apoptosis remain to be determined in the future.
HIPKs
Homeodomain-interacting protein kinases (HIPKs) belong to a novel family of predominantly nuclear serine/threonine kinases. In humans and mice the family comprises three members, HIPK1-3, which were originally identified as corepressors for homeodomain transcription factors.89 HIPKs belong to the dual-specificity tyrosine phosphorylation-regulated kinase family of protein kinases90 and function in transcriptional regulation,91,92 growth suppression93 and apoptosis.24,25 HIPK2, which mediates p53 phosphorylation and ultraviolet (UV)-induced apoptosis24,25 (for details, see section on ‘Regulation of p53 activity in PML-NBs’) was found to interact with TRADD when overexpressed in 293T cells.94 Whether this interaction is of importance for the TRADD-or TRADD-DD-induced cell killing remains to be determined in the future. Murine HIPK3/FIST (Fas-interacting signal transducer), a close relative of HIPK2, has been identified by its interaction with the intracellular domain of CD95 in a yeast two-hybrid screen. Interestingly, ectopically expressed HIPK3 also coprecipitates with FADD and Daxx and a fraction of HIPK3 is found in PML-NBs.95 Overexpression of HIPK3 induces FADD phosphorylation and inhibits CD95-induced c-Jun N-terminal kinase (JNK) activation. However, HIPK3 expression showed no effect on CD95-induced apoptosis in the cell system used. Unexpectedly, FADD was recently found to localise preferentially to the nucleus in different adherent cell lines96 and, additionally, murine FADD was demonstrated to be essential for regulating cell proliferation and cell cycle progression in lymphocytes.97 The latter function depends on its specific phosphorylation at serine, 191,97 a conserved site equivalent to serine 194 in human FADD.98 FADD-deficient mice die in utero, suggesting that FADD also plays a role in nonlymphoid tissue proliferation.99 Future studies are required to reveal a more detailed view about the function of HIPKs in death receptor signalling and their role in FADD phosphorylation.
Daxx
Daxx is a multifunctional protein that participates in different apoptosis pathways.100,101,102,103 The protein was originally identified by its interaction with the intracellular domain of CD95 in a yeast two-hybrid screen and described to act as an adaptor protein that activates the MAP3K apoptosis signal-regulating kinase 1 (ASK1) and triggers the activation of the JNK signalling pathway and apoptosis.100,101 A recent report suggests that the CD95–Daxx–ASK1 signalling route in particular operates in a cell type-specific manner during apoptosis of motoneurons.104 In addition to its role in the cytoplasm, Daxx also fulfils an important function in the nucleus in transcriptional regulation. Dependent on the cellular and the molecular context, Daxx acts as a transcriptional corepressor8,105,106,107 or coactivator.108 Several laboratories demonstrated that Daxx localises to the nucleus where it is found in association with chromatin, centromeres and PML-NBs.8,105,106,107,109 The subcellular localisation of Daxx can be regulated by its interaction with the kinase ASK1, which recruits Daxx independent of its kinase activity to the cytoplasm.110,111 In the cytoplasm, Daxx and ASK1 can initiate a caspase-independent apoptosis pathway that is inhibited by the overexpression of the small heat-shock protein Hsp27.112 Recently, Daxx was shown to function, similar to its role in CD95 signalling, as an adaptor for the transforming growth factor (TGF)-β type II receptor in order to transduce TGF-β-induced JNK activation and apoptosis in hepatocytes and B cells.113 In this respect, it is interesting to note that HIPK2 interacts with Daxx and releases it from PML-NBs by inducing their disintegration. HIPK2 cooperates with Daxx in TGF-β-induced JNK activation and apoptosis in human hepatoma cells (Hofmann TG, Stollberg N, Schmitz ML, Will H, manuscript submitted). Recently, Daxx has also been demonstrated to interact with other members of the HIPK family of proteins,95,114 which suggests a redundant function of HIPKs in regulating the subcellular localisation and the function of Daxx.
Daxx interacts with SUMOlated PML8,37 and both proteins cooperate in a nuclear apoptosis pathway in activation-induced cell death in splenocytes that depends on PML expression.102 These findings indicate an important role of SUMO-1 in apoptosis regulation. In addition, a proapoptotic function of Daxx has also been found in human B progenitor cells, where Daxx is upregulated by interferon treatment and regulates lymphopoiesis through downregulation of the antiapoptotic regulator Bcl-2 followed by the induction of apoptosis.103
In contrast to its proapoptotic functions, Daxx also exerts an antiapoptotic function during embryogenesis.115 Moreover, recent work using siRNA-mediated downregulation of Daxx also demonstrated an antiapoptotic function of Daxx in different cell lines, demonstrating that this effect is not just restricted to embryonic development.116 Future studies are needed to clarify the discrepancies in the role of Daxx in apoptosis, in particular considering a possible cell type-specific function, which might in part be explained by its molecular context-dependent gene repressive or stimulatory function.108 Since Daxx also interacts with p53,117 it would be interesting to study whether the apoptosis in Daxx-deficient mice depends on p53, and whether Daxx might serve as a corepressor for unrestrained p53 activation during development. In this respect, it is interesting to note that Daxx can repress the transactivating potential of the transcription factor Ets-1,106 which in turn appears to be essential for stress-induced p53 target gene expression as revealed by experiments with Ets-1-deficient embryonic stem cells.118
Regulating p53-dependent apoptosis from the PML-NBs
In addition to their regulatory function in receptor-mediated apoptosis pathways, PML and its associated NBs also regulate the activity of a central apoptotic regulator – the tumour suppressor protein p53.
p53 activation and regulation
The tumour suppressor protein p53 is a cellular key player controlling cell cycle arrest, premature cellular senescence and apoptosis.119 p53 is a modular organised transcription factor, comprising an N-terminal transactivation domain, a central DNA-binding domain and a C-terminal regulatory domain. p53 activation is mainly regulated by its subcellular localisation and on the post-transcriptional level through protein stabilisation and post-translational modification. In normal dividing cells, p53 is kept silent at almost undetectable protein levels through its association with the proto-oncoprotein Mdm2 (or its human homologue Hdm2). Mdm2, a RING finger E3 ubiquitin ligase, binds p53 and mediates its ubiquitination that results in proteasomal degradation of p53.120,121 In response to a plethora of different stimuli – including genotoxic insult (UV- or γ-irradiation), chemotherapeutics, hypoxia, interference with DNA synthesis by stalling replication forks, oncogenic transformation and replicative senescence – p53 gets stabilised via abolishing its proteolytic degradation.122,123 p53 mainly acts as a sequence-specific transcription factor that transactivates or represses its multiple target genes, thereby controlling its effector pathways either leading to cell cycle arrest, premature cellular senescence or apoptosis. However, p53 can also induce apoptosis via transcription-independent mechanisms and translocation to the mitochondria triggering cytochrome c release and caspase activation.124,125,126,127
Basically, two mechanistically different pathways mediating p53 activation have been identified. One pathway relies on the activation of a set of protein kinases, which directly phosphorylate serine or threonine residues within the N-terminal transactivation domain and the C-terminal regulatory domain of p53.128 For example, the kinases ataxia-telangiectasia mutated (ATM) and Chk2 are activated in response to DNA damage induced by UV- or γ-irradiation and mediate the phosphorylation of Serine 15, which triggers disruption of the Mdm2-p53 complex resulting in an increased p53 half-life.129,130 The second mechanism, which operates during deregulated expression of cellular or viral oncogenes, leads to a phosphorylation-independent stabilisation of p53 involving the INK4a locus encoded protein p14ARF. p14ARF – a small protein transcriptionally regulated by the kinase DAPK131 and the transcription factors and proto-oncogenes E2F-1 and c-myc – binds to MDM2 and inhibits its ubiquitin ligase activity resulting in p53 stabilisation and target gene expression.132 Thus, hyperproliferative stimuli through the expression of cellular oncogenes, such as oncogenic RasV12, c-Myc or E2F-1, induce upregulation of p14ARF and trigger a cellular tumour suppressive response that counteracts transformation. In primary cells, the resulting p53 activation triggers premature cellular senescence – a permanent, irreversible cell cycle arrest in the G1 phase.133
The different p53 activation mechanisms converge on the level of p53 stabilisation, which is regulated by p53 post-translational modification pattern – including acetylation, phosphorylation, deubiquitination and SUMO-1 conjugation. Several recent findings argue for an important role of PML-NBs and, their components in the control of p53s post-translational modification and activity.
Regulation of p53 activity in PML-NBs
Accumulating experimental evidence provided by several independent groups indicates a role of the PML-NB and its components in p53 activation in response to different stimuli. Besides p53 itself, multiple factors regulating p53 are found within or in association with PML-NBs, in particular PML,15,22,23 CBP,14 Hdm21,34 HIPK2,24,25 Chk2,135,136 hSir217 and herpesvirus-associated ubiquitin-specific protease (HAUSP).137,138 This significant accumulation of factors involved in p53 regulation raises the assumption that PML-NBs form supramolecular scaffolds that establish a peculiar molecular microenvironment enabling efficient post-translational modification of a fraction of p53. Similar to most PML-NB components, p53 is not a constitutive resident and its recruitment to PML-NBs is mediated by all PML isoforms,16 but only interaction with PML IV regulates p53 activity.22,23 Genotoxic stress induces PML-NB formation and leads to p53-dependent apoptosis induction, which in part requires PML.22,23 Upon high-dose UV-radiation, HIPK2 localises together with p53 and CBP in PML-NBs and phosphorylates p53 at serine 46.24,25 HIPK2 expression results in p53-dependent apoptosis that is associated both with p53 Ser46 phosphorylation and Lys382 acetylation,24,25 modifications previously shown to trigger p53 activation and apoptosis.139,140,141,142 A recent publication suggests that the nucleoplasmic fraction of PML IV mediates the effect on p53 Ser46 phosphorylation and Lys382 acetylation during premature senescence, indicating that the PML-p53 interaction can also take place in the nucleoplasm.16 Although these experiments show that PML-NBs are not required for Ser46 phosphorylation, they do not exclude the fact that PML-NBs might favour p53 modification and promote its activation before its release into the nucleoplasm and to its target promoters at the chromatin. PML is required for HIPK2-mediated p53 Ser46 phosphorylation and can be restored in PML−/− cells by ectopically expressed PML IV (Möller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Dröge W, Will H, Schmitz ML, manuscript submitted). In addition, the p53 Ser20 kinase Chk2 was recently identified in PML-NBs in response to genotoxic stress.135 The overexpression of PML prolonged the period of p53 Ser20 phosphorylation and protected it against mdm2-mediated ubiquitination and degradation, thus leading to increased p53 stability and activity.135
Several groups reported that p53 is a target protein for SUMO-1.42,43,143,144 SUMO-1 is covalently attached to specific lysine residues of its target proteins by an enzymatic machinery, which is similar to that of ubiquitin modification.45 p53 is SUMOlated at Lys386 within its regulatory domain through a complex containing the SUMO-1 conjugating enzyme Ubc9 and the SUMO-1 ligase protein inhibitor of activated STAT (PIAS)1.51,144 However, addressing the function of p53 SUMOlation on p53 activity led to contradictory results, reaching from increased activity,42,43 no effect143 to decreased transcriptional activity of p53.144 The reason for these discrepancies should be clarified in the future, although it is currently not clear whether SUMOlation of p53 might take place in PML-NBs. Interestingly, similar to SUMO-1, PIAS proteins also appear to localise to PML-NBs.52 Recently, the deubiquitinating enzyme HAUSP, which colocalises with PML-NBs,137 was found to stabilise p53 and to activate its proapoptotic activity.138 Whether HAUSP can exert its function in PML-NBs remains to be studied.
Besides several p53-activating enzymes, factors implicated in negative regulation of p53 were also found in PML-NBs. For example, the evolutionarily conserved NAD-dependent histone deacetylase hSir2 localises to PML-NBs.17 hSir2 inhibits p53 activity through deacetylation of Lys382 and antagonises genotoxic stress-induced apoptosis.145,146 The localisation of CBP and hSir2 to PML-NBs, two factors that exert antagonistic functions on p53 activity, suggest a potential mechanism for the regulation of p53 function in PML-NBs the change of its acetylation status, depending on the molecular context and stimulus. In addition, a fraction of the p53 degrading E3 ubiquitin ligase Hdm2 (human Mdm2) was found associated with PML-NBs.134 Although the functional meaning of this finding is currently not clear, this may indicate that PML-NBs can regulate p53 ubiquitination and proteasome-dependent degradation. If true, this would be another level of PML-NB-mediated regulation of apoptosis.
Taken together, there is accumulating evidence for the model that a fraction of p53 is post-translationally modified within the PML-NBs under specific cellular conditions (Figure 3). PML-NBs might function preferentially by serving as scaffolds to increase the local concentrations of factors implicated in p53 post-translational modification, including site-specific acetylation and phosphorylation. Subsequently, modified p53 may be released from PML-NBs and targeted to its site of action at its target gene promoters. Recent findings that SUMO-specific protease SuPr-154 and HIPK2 induce PML-NB disintegration147 (Hofmann TG, Stollberg N, Schmitz ML, Will H, manuscript submitted), suggest an attractive molecular mechanism of how modified p53 and other factors can be released from PML-NBs. The specific molecular context of the multiple positive and negative regulators of p53 concentrated in PML-NBs may lead to stabilisation, activation, inactivation or even degradation of this cellular key protein. Answers to these many open questions are likely to contribute to a better understanding of the role of p53 and PML-NBs in human diseases and cancer.

Speculative model of PML-NB components and their role in p53 regulation. Upon oncogenic transformation-induced cellular senescence, UV-induced stress and treatment with the chemotherapeutic drug arsenic trioxide (As2O3) PML mediates corecruitment of p53 and different p53 regulating factors to PML-NBs, where they colocalise with p53. p53 is subsequently differentially post-translationally modified and activated. Active, modified p53 leaves the PML-NBs and is targeted to the chromatin where it regulates the expression of its specific target genes, thereby triggering the different p53-effector pathways. The different post-translational modifications on p53 affected in these processes are indicated. S, SUMOlation, P, phosphorylation, Ac, acetylation, Ub, ubiquitination. For a detailed description, see text
Summary
Several independent lines of evidence point to an important function of PML-NBs and their components in regulating extrinsic and intrinsic apoptosis pathways, in particular those elicited by ligation of death-inducing cell surface receptors or those resulting in p53 activation. First, data from PML−/− cells show that these cells have multiple defects in different apoptosis pathways, including death receptor-mediated ones and p53-dependent pathways. Second, APL blasts that express the PML-RARα oncoprotein and have disrupted PML-NBs are resistant towards various apoptosis stimuli, and As2O3 treatment induces reformation of PML-NBs accompanied by apoptosis of the blasts. Third, there is evidence that signalling molecules participating in receptor-mediated apoptosis pathways (Daxx, TRADD) at a receptor proximal level localise to PML-NBs and also participate in apoptosis regulation employing nuclear pathways. This suggests a regulatory function of PML-NBs in these pathways by regulating the bioavailability of specific apoptotic signal transducers. Strikingly, a significant number of proteins that fulfil an important function in regulating p53 activity (CBP, PML, HIPK2, HAUSP, hSir2, Chk2) are recruited along with p53 to PML-NBs in response to genotoxic insult or cellular senescence, and regulate p53 effector function in a PML-dependent manner. Even more striking, multiple important post-translational modifications associated with p53 stabilisation and activation, including Ser15, Ser20 and Ser46 phosphorylation as well as Lys382 acetylation, are dependent on the presence of PML, highlighting the emerging role of these nuclear domains in p53 regulation.
Concluding remarks and future perspectives
The mechanism by which PML-NBs and their components regulate the function of their components that participate in apoptosis signal transduction awaits further elucidation. Current knowledge about the function of PML and its NB components in apoptosis regulation suggests an intricate network between different apoptosis pathways and PML-NBs. As discussed, PML-NBs may modulate death receptor pathways by controlling the bioavailability and the quality of apoptogenic PML-NB components that operate in these pathways. Therefore, it will be important to know whether other factors involved in receptor-mediated apoptosis signalling or p53 regulation localise to PML-NBs, and if so, how their localisation affects the sensitivity towards different apoptosis pathways.
Although currently available evidence strongly suggests that PML-NBs represent nuclear post-translational modification platforms for p53, one important question to be answered in the future is whether the PML-NB-associated fractions of HIPK2, CBP, hSir2, HAUSP, Chk2 and/or the nucleoplasmic fractions thereof are catalytically active on p53. In order to catch a molecular glimpse of how PML-NBs are coupled to various apoptosis pathways, we still need the answer to a couple of open questions. At which level and how does PML act in death receptor and other apoptosis pathways? What are the functions of cytoplasmic and nuclear PML isoforms? Which are the players in the PML- and TRADD-induced caspase-independent apoptosis pathways? Does PML also regulate apoptosis pathways mediated by other death receptors? These and other related important questions will probably be answered in the near future and will help to shed light on the molecular function of the PML-NBs.
Abbreviations
- AIF:
-
apoptosis-inducing factor
- APAF-1:
-
apoptosis-activating factor 1
- APL:
-
acute promyelocytic leukaemia
- ARF:
-
alternative reading frame
- As2O3:
-
arsenic trioxide
- ASK1:
-
apoptosis signal-regulating kinase 1
- ATM:
-
ataxia-telangiectasia mutated
- CBP:
-
CREB-binding protein
- DD:
-
death domain
- DED:
-
death-effector domain
- DISC:
-
death-inducing signalling complex
- HAUSP:
-
herpesvirus-associated ubiquitin-specific protease
- HIPK:
-
homeodomain-interacting protein kinase
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- PIAS:
-
protein inhibitor of activated STAT
- PML:
-
promyelocytic leukaemia
- SAE:
-
SUMO-1-activating enzyme
- SUMO:
-
small ubiquitin-related modifier
- TGF:
-
transforming growth factor
- TNF-R:
-
tumour necrosis factor receptor
- TRADD:
-
TNF-R1-associated death domain protein
- TRAIL:
-
tumour necrosis factor-related apoptosis-inducing ligand
- UV:
-
ultraviolet
References
Spector DL (2001) Nuclear domains. J. Cell Sci. 114: 2891–2893
Borden KL (2002) Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol. Cell. Biol. 22: 5259–5269
Dyck JA, Maul GG, Miller WJ, Chen JD, Kakizuka A and Evans RM (1994) A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell 76: 333–343
Weis K, Rambaud S, Lavau C, Jansen J, Carvalho T, Carmo-Fonseca M, Lamond A and Dejean A (1994) Retinoic acid regulates aberrant nuclear localization of PML-RAR alpha in acute promyelocytic leukemia cells. Cell 76: 345–356
Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P and de The H (2001) Role of promyelocytic leukemia (PML) SUMOlation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 193: 1361–1372
Muratani M, Gerlich D, Janicki SM, Gebhard M, Eils R and Spector DL (2002) Metabolic-energy-dependent movement of PML bodies within the mammalian cell nucleus. Nat. Cell Biol. 4: 106–110
Wiesmeijer K, Molenaar C, Bekeer IM, Tanke HJ and Dirks RW (2002) Mobile foci of Sp100 do not contain PML: PML bodies are immobile but PML and Sp100 proteins are not. J. Struct. Biol. 140: 180–188
Li H, Leo C, Zhu J, Wu X, O'Neil J, Park EJ and Chen JD (2000) Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 20: 1784–1796
Zhong S, Salomoni P and Pandolfi PP (2000) The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2: E85–E90
Fagioli M, Alcalay M, Tomassoni L, Ferrucci PF, Mencarelli A, Riganelli D, Grignani F, Pozzan T, Nicoletti I, Grignani F and Pelicci PG (1998) Cooperation between the RING + B1–B2 and coiled-coil domains of PML is necessary for its effects on cell survival. Oncogene 16: 2905–2913
Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S and Borden KL (2001) PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. EMBO J. 20: 4547–4559
Everett RD (2001) DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20: 7266–7273
Regad T and Chelbi-Alix MK (2001) Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20: 7274–7286
Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP and Pelicci PG (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406: 207–210
Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C and Lowe SW (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 14: 2015–2027
Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG and Dejean A (2002) Deconstructing PML-induced premature senescence. EMBO J. 21: 3358–3369
Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG and Kouzarides T (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21: 2383–2396
Salomoni P and Pandolfi PP (2002) The role of PML in tumor suppression. Cell 108: 165–170
Bischof O, Kim SH, Irving J, Beresten S, Ellis NA and Campisi J (2001) Regulation and localization of the bloom syndrome protein in response to DNA damage. J. Cell Biol. 153: 367–380
Carbone R, Pearson M, Minucci S and Pelicci PG (2002) PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 21: 1633–1640
Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R and Pandolfi PP (1998) PML is essential for multiple apoptotic pathways. Nat. Genet. 20: 266–272
Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C and Del Sal G (2000) Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 19: 6185–6195
Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W and Pandolfi PP (2000) The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2: 730–736
D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E and Soddu S (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 4: 11–19
Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H and Schmitz ML (2002) Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 4: 1–10
Zhu J, Lallemand-Breitenbach V and de The H (2001) Pathways of retinoic acid- or arsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradation in disease remission. Oncogene 20: 7257–7265
de Thé H, Chomienne C, Lanotte M, Degos L and Dejean A (1990) The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 347: 558–561
Borrow J, Goddard AD, Sheer D and Solomon E (1990) Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science 249: 1577–1580
Goddard AD, Borrow J, Freemont PS and Solomon E (1991) Characterization of a zinc finger gene disrupted by the t(15;17) in acute promyelocytic leukemia. Science 254: 1371–1374
Kakizuka A, Miller Jr WH, Umesono K, Warrell Jr RP, Frankel SR, Murty VV, Dmitrovsky E and Evans RM (1991) Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 66: 663–674
Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Shinagawa T, Ichikawa-Iwata E, Zhong S, Pandolfi PP and Ishii S (2001) Role of pml and pml-raralpha in mad-mediated transcriptional repression. Mol. Cell 7: 1233–1243
Zhu J, Chen Z, Lallemand-Breitenbach V and de The H (2002) How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2: 705–713
Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S and Pelicci PG (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295: 1079–1082
Le XF, Yang P and Chang KS (1996) Analysis of the growth and transformation suppressor domains of promyelocytic leukemia gene, PML. J. Biol. Chem. 271: 130–135
Saurin AJ, Borden KLB, Boddy MN and Freemont PS (1996) Does this have a familiar RING? Trends Biochem. Sci. 21: 208–214
Jensen K, Shiels C and Freemont PS (2001) PML protein isoforms and the RBCC/TRIM motif. Oncogene 20: 7223–7233
Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss Jr JF and Maul GG (1999) PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147: 221–234
Zhong S, Müller S, Ronchetti S, Freemont PS, Dejean A and Pandolfi PP (2000) Role of SUMO-1-modified PML in nuclear body formation. Blood 95: 2748–2752
Lai HK and Borden KL (2000) The promyelocytic leukemia (PML) protein suppresses cyclin D1 protein production by altering the nuclear cytoplasmic distribution of cyclin D1 mRNA. Oncogene 19: 1623–1634
Sternsdorf T, Jensen K and Will H (1997) Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 139: 1621–1634
Jang MS, Ryu SW and Kim E (2002) Modification of Daxx by small ubiquitin-related modifier-1. Biochem. Biophys. Res. Commun. 295: 495–500
Gostissa M, Hengstermann A, Fogal V, Sandy P, Schwarz SE, Scheffner M and Del Sal G (1999) Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 18: 6462–6471
Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP and Hay RT (1999) SUMO-1 modification activates the transcriptional response of p53. EMBO J. 18: 6455–6461
Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia-Wilson E, Perkins ND and Hay RT (2003) p300 transcriptional repression is mediated by SUMO modification. Mol. Cell 11: 1043–1054
Melchior F (2000) SUMO – nonclassical ubiquitin. Annu. Rev. Cell. Dev. Biol. 16: 591–626
Desterro JM, Rodriguez MS, Kemp GD and Hay RT (1999) Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 274: 10618–10624
Desterro JM, Thomson J and Hay RT (1997) Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 417: 297–300
Johnson ES and Blobel G (1997) Ubc9p is the conjugating enzyme for the ubiquitin-like protein smt3p. J. Biol. Chem. 272: 26799–26802
Kagey MH, Melhuish TA and Wotton D (2003) The Polycomb Protein Pc2 Is a SUMO E3. Cell 113: 127–137
Pichler A, Gast A, Seeler JS, Dejean A and Melchior F (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108: 109–120
Kahyo T, Nishida T and Yasuda H (2001) Involvement of PIAS1 in the SUMOlation of tumor suppressor p53. Mol. Cell 8: 713–718
Sachdev S, Bruhn L, Sieber H, Pichler A, Melchior F and Grosschedl R (2001) PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15: 3088–3103
Li H and Chen JD (2000) PML and the oncogenic nuclear domains in regulating transcriptional repression. Curr. Opin. Cell Biol. 12: 641–644
Best JL, Ganiatsas S, Agarwal S, Changou A, Salomoni P, Shirihai O, Meluh PB, Pandolfi PP and Zon LI (2002) SUMO-1 protease-1 regulates gene transcription through PML. Mol. Cell 10: 843–855
Sapetschnig A, Rischitor G, Braun H, Doll A, Schergaut M, Melchior F and Suske G (2002) Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO J. 21: 5206–5215
Matunis MJ (2002) On the road to repair: PCNA encounters SUMO and ubiquitin modifications. Mol. Cell 10: 441–442
Jackson PK (2001) A new RING for SUMO: wrestling transcriptional responses into nuclear bodies with PIAS family E3 SUMO ligases. Genes Dev. 15: 3053–3058
Vaux DL, Haecker G and Strasser A (1994) An evolutionary perspective on apoptosis. Cell 76: 777–779
Ameisen JC (2002) On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 9: 367–393
Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462
Krammer PH (2000) CD95's deadly mission in the immune system. Nature 407: 789–795
Ross CA (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35: 819–822
Igney FH and Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2: 277–288
Strasser A, O'Connor L and Dixit VM (2000) Apoptosis signaling. Annu. Rev. Biochem. 69: 217–245
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W and Vandenabeele P (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 188: 919–930
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W and Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187: 1477–1485
Salvesen GS and Dixit VM (1999) Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. USA 96: 10964–10970
Puthalakath H and Strasser A (2002) Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9: 505–512
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ and Ashkenazi A (2000) Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12: 611–620
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH and Peter ME (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14: 5579–5588
Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH and Peter ME (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 16: 2794–2804
Fischer U, Janicke RU and Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell. Death Differ. 10: 76–100
Rokudai S, Fujita N, Kitahara O, Nakamura Y and Tsuruo T (2002) Involvement of FKHR-dependent TRADD expression in chemotherapeutic drug-induced apoptosis. Mol. Cell. Biol. 22: 8695–8708
Dijkers PF, Medema RH, Lammers JW, Koenderman L and Coffer PJ (2000) Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 10: 1201–1204
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ and El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 4: 842–849
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694
Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7: 673–682
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M and Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188: 2033–2045
Tanikawa C, Matsuda K, Fukuda S, Nakamura Y and Arakawa H (2003) p53RDL1 regulates p53-dependent apoptosis. Nat. Cell Biol. 5: 216–223
Liu ZG, Hsu H, Goeddel DV and Karin M (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87: 565–576
Van Antwerp DJ, Martin SJ, Kafri T, Green DR and Verma IM (1996) Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274: 787–789
Wang CY, Mayo MW and Baldwin Jr AS (1996) TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274: 784–787
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS and Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221
Altucci L, Rossin A, Raffelsberger W, Reitmair A, Chomienne C and Gronemeyer H (2001) Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 7: 680–606
Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC and de Thé H (1998) PML induces a novel caspase-independent death process. Nat. Genet. 20: 259–265
Morgan M, Thorburn J, Pandolfi PP and Thorburn A (2002) Nuclear and cytoplasmic shuttling of TRADD induces apoptosis via different mechanisms. J. Cell Biol. 157: 975–984
Hsu H, Xiong J and Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81: 495–504
Kim YH, Choi CY, Lee SJ, Conti MA and Kim Y (1998) Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J. Biol. Chem. 273: 25875–25879
Hofmann TG, Mincheva A, Lichter P, Droge W and Lienhard Schmitz M (2000) Human homeodomain-interacting protein kinase-2 (HIPK2) is a member of the DYRK family of protein kinases and maps to chromosome 7q32–q34. Biochimie 82: 1123–1127
Choi CY, Kim YH, Kwon HJ and Kim Y (1999) The homeodomain protein NK-3 recruits Groucho and a histone deacetylase complex to repress transcription. J. Biol. Chem. 274: 33194–33197
Moilanen AM, Karvonen U, Poukka H, Janne OA and Palvimo JJ (1998) Activation of androgen receptor function by a novel nuclear protein kinase. Mol. Biol. Cell. 9: 2527–2543
Pierantoni GM, Fedele M, Pentimalli F, Benvenuto G, Pero R, Viglietto G, Santoro M, Chiariotti L and Fusco A (2001) High mobility group I (Y) proteins bind HIPK2, a serine-threonine kinase protein which inhibits cell growth. Oncogene 20: 6132–6341
Li X, Wang Y, Debatin KM and Hug H (2000) The serine/threonine kinase HIPK2 interacts with TRADD, but not with CD95 or TNF-R1 in 293T cells. Biochem. Biophys. Res. Commun. 277: 513–517
Rochat-Steiner V, Becker K, Micheau O, Schneider P, Burns K and Tschopp J (2000) FIST/HIPK3: a Fas/FADD-interacting serine/threonine kinase that induces FADD phosphorylation and inhibits fas-mediated Jun NH(2)-terminal kinase activation. J. Exp. Med. 192: 1165–1174
Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, Clarke AR and Frisch SM (2003) Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 100: 5211–5216
Hua ZC, Sohn SJ, Kang C, Cado D and Winoto A (2003) A function of fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity 18: 513–521
Scaffidi C, Volkland J, Blomberg I, Hoffmann I, Krammer PH and Peter ME (2000) Phosphorylation of FADD/ MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. J. Immunol. 164: 1236–1242
Zhang J, Cado D, Chen A, Kabra NH and Winoto A (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/MORT1. Nature 392: 296–300
Yang X, Khosravi-Far R, Chang HY and Baltimore D (1997) Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89: 1067–1076
Chang HY, Nishitoh H, Yang X, Ichijo H and Baltimore D (1998) Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281: 1860–1863
Zhong S, Salomoni P, Ronchetti S, Guo A, Ruggero D and Pandolfi PP (2000) Promyelocytic leukemia protein (PML) and Daxx participate in a novel nuclear pathway for apoptosis. J. Exp. Med. 191: 631–640
Gongora R, Stephan RP, Zhang Z and Cooper MD (2001) An essential role for Daxx in the inhibition of B lymphopoiesis by type I interferons. Immunity 14: 727–737
Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G and Pettmann B (2002) Motoneuron death triggered by a specific pathway downstream of Fas. potentiation by ALS-linked SOD1 mutations. Neuron 35: 1067–1083
Torii S, Egan DA, Evans RA and Reed JC (1999) Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J. 18: 6037–6049
Li R, Pei H, Watson DK and Papas TS (2000) EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene 19: 745–753
Lehembre F, Muller S, Pandolfi PP and Dejean A (2001) Regulation of Pax3 transcriptional activity by SUMO-1-modified PML. Oncogene 20: 1–9
Emelyanov AV, Kovac CR, Sepulveda MA and Birshtein BK (2002) The interaction of Pax5 (BSAP) with Daxx can result in transcriptional activation in B cells. J. Biol. Chem. 277: 11156–11164
Pluta AF, Earnshaw WC and Goldberg IG (1998) Interphase-specific association of intrinsic centromere protein CENP-C with hDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 111: 2029–2041
Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ and Kim S (2001) Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. J. Biol. Chem. 276: 39103–39106
Charette SJ, Lambert H and Landry J (2001) A kinase-independent function of Ask1 in caspase-independent cell death. J. Biol. Chem. 276: 36071–36074
Charette SJ and Landry J (2000) The interaction of HSP27 with Daxx identifies a potential regulatory role of HSP27 in Fas-induced apoptosis. Ann. NY Acad. Sci. 926: 126–131
Perlman R, Schiemann WP, Brooks MW, Lodish HF and Weinberg RA (2001) TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 3: 708–714
Ecsedy JA, Michaelson JS and Leder P (2003) Homeodomain-interacting protein kinase 1 modulates daxx localization, phosphorylation, and transcriptional activity. Mol. Cell. Biol. 23: 950–960
Michaelson JS, Bader D, Kuo F, Kozak C and Leder P (1999) Loss of daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 13: 1918–1923
Michaelson JS and Leder P (2003) RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 116: 345–352
Ohiro Y, Usheva A, Kobayashi S, Duffy SL, Nantz R, Gius D and Horikoshi N (2003) Inhibition of stress-inducible kinase pathways by tumorigenic mutant p53. Mol. Cell. Biol. 23: 322–334
Xu D, Wilson TJ, Chan D, De Luca E, Zhou J, Hertzog PJ and Kola I (2002) Ets1 is required for p53 transcriptional activity in UV-induced apoptosis in embryonic stem cells. EMBO J. 21: 4081–4093
Vogelstein B, Lane D and Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310
Kubbutat MH, Jones SN and Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303
Haupt Y, Maya R, Kazaz A and Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299
Lundberg AS, Hahn WC, Gupta P and Weinberg RA (2000) Genes involved in senescence and immortalization. Curr. Opin. Cell Biol. 12: 705–709
Oren M (1999) Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 274: 36031–36034
Caelles C, Helmberg A and Karin M (1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370: 220–223
Haupt Y, Rowan S, Shaulian E, Vousden KH and Oren M (1995) Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 9: 2170–2183
Marchenko ND, Zaika A and Moll UM (2000) Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J. Biol. Chem. 275: 16202–16212
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P and Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 11: 577–590
Wahl GM and Carr AM (2001) The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat. Cell Biol. 3: E277–E286
Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E and Kastan MB (1997) DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11: 3471–3481
Shieh SY, Ikeda M, Taya Y and Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334
Raveh T, Droguett G, Horwitz MS, DePinho RA and Kimchi A (2001) DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 3: 1–7
Sherr CJ (2001) The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2: 731–737
Campisi J (2001) Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 11: S27–S31
Lain S, Midgley C, Sparks A, Lane EB and Lane DP (1999) An inhibitor of nuclear export activates the p53 response and induces the localization of HDM2 and p53 to U1A-positive nuclear bodies associated with the PODs. Exp. Cell Res. 248: 457–472
Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP and Haupt Y (2003) PML protects p53 from Mdm2-mediated inhibition and degradation. J. Biol. Chem. 16: 16
Yang S, Kuo C, Bisi JE and Kim MK (2002) PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat. Cell Biol. 4: 865–870
Everett RD, Meredith M, Orr A, Cross A, Kathoria M and Parkinson J (1997) A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16: 566–577
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J and Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416: 648–653
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y and Taya Y (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862
Gu W, Shi XL and Roeder RG (1997) Synergistic activation of transcription by CBP and p53. Nature 387: 819–823
Lill NL, Grossman SR, Ginsberg D, DeCaprio J and Livingston DM (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature 387: 823–827
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS and Kelly K (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89: 1175–1184
Kwek SS, Derry J, Tyner AL, Shen Z and Gudkov AV (2001) Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene 20: 2587–2599
Schmidt D and Muller S (2002) Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl. Acad. Sci. USA 99: 2872–2877
Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L and Weinberg RA (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159
Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L and Gu W (2001) Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107: 137–148
Engelhardt OG, Boutell C, Orr A, Ullrich E, Haller O and Everett RD (2003) The homeodomain-interacting kinase PKM (HIPK-2) modifies ND10 through both its kinase domain and a SUMO-1 interaction motif and alters the posttranslational modification of PML. Exp. Cell Res. 283: 36–50
Wu WS, Xu ZX, Hittelman WN, Salomoni P, Pandolfi PP and Chang KS (2003) Promyelocytic leukemia protein sensitizes tumor necrosis factor alpha-induced apoptosis by inhibiting the NF-kappaB survival pathway. J. Biol. Chem. 278: 12294–12304
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft, the Deutsche Krebshilfe, the BMBF and the ‘Stiftung zur Bekämpfung Neuroviraler Erkrankungen’. The Heinrich-Pette-Institut is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G. Mellino
Rights and permissions
About this article
Cite this article
Hofmann, T., Will, H. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ 10, 1290–1299 (2003). https://doi.org/10.1038/sj.cdd.4401313
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401313
Keywords
This article is cited by
-
Multipronged regulation of autophagy and apoptosis: emerging role of TRIM proteins
Cellular & Molecular Biology Letters (2024)
-
The interactions between PML nuclear bodies and small and medium size DNA viruses
Virology Journal (2023)
-
Increased nuclear permeability is a driver for age-related motoneuron loss
GeroScience (2020)
-
Regulation of the oncogenic phenotype by the nuclear body protein ZC3H8
BMC Cancer (2018)
-
RanBP2 regulates the anti-retroviral activity of TRIM5α by SUMOylation at a predicted phosphorylated SUMOylation motif
Communications Biology (2018)
