
Abstract
In the present study, we demonstrate that Ca2+-induced growth inhibition and induction of differentiation in a line of human colon carcinoma cells (CBS) is dependent on mitogen-activated protein (MAP) kinase signaling and is associated with upregulation of extracellular calcium-sensing receptor (CaSR) expression. When CBS cells were grown in Ca2+-free medium and then switched to medium supplemented with 1.4 mM Ca2+, proliferation was reduced and morphologic features of differentiation were expressed. E-cadherin, which was minimally expressed in nonsupplemented medium, was rapidly induced in response to Ca2+ stimulation. Sustained activation of the extracellular signal-regulated kinase (ERK) occured in Ca2+-supplemented medium. When an inhibitor of ERK activation (10 μ M U0126) was included in the Ca2+-supplemented culture medium, ERK-activation did not occur. Concomitantly, E-cadherin was not induced, cell proliferation remained high and differentiation was not observed. The same level of Ca2+ supplementation that induced MAP kinase activation also stimulated CaSR upregulation in CBS cells. A clonal isolate of the CBS line that did not upregulate CaSR expression in response to extracellular Ca2+ was isolated from the parent cells. This isolate failed to produce E-cadherin or undergo growth inhibition/induction of differentiation when exposed to Ca2+ in the culture medium. However, ERK-activation occurred as efficiently in this isolate as in parent CBS cells or in a cloned isolate that underwent growth reduction and differentiation in response to Ca2+ stimulation. Together, these data indicate that CaSR upregulation and MAP kinase signalling are both intermediates in the control of colon carcinoma cell growth and differentiation. They appear to function, at least in part, independently of one another.
Similar content being viewed by others


Main
Calcium (Ca2+) has growth-inhibiting and differentiation-promoting activities for a variety of normal and malignant epithelial cells, including cells of the gastrointestinal tract (Lamprecht and Lipkin, 2003). Compelling experimental and epidemiological evidence suggests an inverse relationship between dietary intake of Ca2+ and risk of colorectal cancer (Sellers et al, 1998; Baron et al, 1999; Lipkin, 1999; Kampman et al, 2000; Wargovich et al, 2000; Wu et al, 2002; McCullough et al, 2003). Although such findings strongly imply a role for extracellular Ca2+ in regulating growth and differentiation in colon epithelium, the mechanism(s) by which this occur(s) is/are poorly understood.
Recent studies suggest that the extracellular calcium-sensing receptor (CaSR) is involved in the regulation of colon carcinoma cell growth and differentiation (Chakrabarty et al, 2003, 2005). The CaSR is expressed in a number of human colon carcinoma cell lines derived from moderately-differentiated tumours. The CaSR promoter has two distinct Ca2+-responsive sites and exposure of colon carcinoma cells to extracellular Ca2+ upregulates CaSR expression. That CaSR expression might be critical to the growth-reduction and differentiation-induction by Ca2+ in these cells is suggested by the finding that gadolinium (Gd3+), an analogue of Ca2+ which binds to the CaSR but does not enter cells (Garrett et al, 1995; Hebert and Brown, 1995; Rutten et al, 1999), is equally efficient as Ca2+ in reducing growth and inducing differentiation (Chakrabarty et al, 2003).
The present studies further our efforts to understand mechanisms of growth regulation in colon epithelium by extracellular Ca2+. Here, we show that the extracellular signal-regulated kinase (ERK) cascade of the mitogen-activated protein (MAP) kinase family is activated in a sustained fashion by extracellular Ca2+ in a line of colon carcinoma cells (CBS), concomitant with growth suppression and induction of differentiation. Interference with ERK activation using the inhibitor U0126 prevents Ca2+-induced alterations in growth and differentiation. A CBS clone that fails to upregulate CaSR in response to Ca2+ also fails to demonstrate growth reduction or induction of differentiation under the same conditions. However, when the CaSR-defective cells are exposed to extracellular Ca2+, ERK activation occurs as efficiently as in the parent cells. Taken together, these data indicate that both CaSR upregulation and MAP kinase signalling are critical intermediates in the control of colon carcinoma cell growth and differentiation. The two events appear to be (at least in part) independent of each other.
Materials and methods
Reagents
Reagents used in these studies included antibodies to the total and phosphorylated forms of ERK, obtained from Cell Signaling Technologies Inc.; Beverly, MA, USA; antibody to E-cadherin, obtained from Chemicon International Inc., Temecula, CA, USA; antibody to CaSR, obtained from Alpha Diagnostic International, San Antonio, TX, USA; and antibody to β-tubulin, obtained from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA. Signalling inhibitors included U0126 (ERK inhibitor), obtained from Cell Signaling Technologies Inc. (Favata et al, 1998); SB203580 (P38 inhibitor), obtained from Calbiochem, San Diego, CA, USA, (Moon et al, 2002); SP600125, an anthrapyrazolone inhibitor of the Jun-N-terminal kinase (JNK) obtained from Axxora LLC, San Diego, CA, USA (Bennett et al, 2001); and LY294002, an inhibitor of phosphatidylinositol-3-kinaase (PI3K) obtained from Cell Signaling Technologies (Panka et al, 2001).
Cell culture
The moderately-differentiated colon carcinoma cell line, CBS, was used in these studies. This cell line was described in our past reports dealing with CaSR expression and colon carcinoma growth control (Chakrabarty et al, 2003, 2005). The cells were maintained in Ca2+-free minimal essential medium – Spinner modified (SMEM) (Sigma Chemical Co., St Louis, MO, USA) supplemented with sodium bicarbonate, amino acids, and 5% foetal bovine serum (FBS). Growth was at 37°C in an atmosphere of 95% air and 5% CO2. Cells were subcultured by brief exposure to trypsin/EDTA solution.
Random cell cloning was used to obtain several isolates of the CBS line. Briefly, a single-cell suspension of the parent cells was fractionated using a flow cytometer (Facscan, Becton-Dickinson) and one cell per well deposited into wells of a 96-well dish containing 100 μl of CBS-conditioned medium. When individual colonies were of sufficient size, they were subcultured into 25-cm2 flasks. When the flasks reached confluence, there were sufficient cells for use in experiments. Cloned isolates were maintained exactly as the parent cells.
Cell proliferation assay
Cells were plated in 24-well tissue culture dishes at 4 × 104 cells per well using Ca2+-free SMEM with 5% FBS as culture medium. After allowing the cells to attach overnight, they were washed twice with Ca2+-free SMEM without FBS and then incubated in Ca2+-free SMEM with 5% dialysed FBS. Different amounts of Ca2+ were then added to individual wells as indicated in the Results. Proliferation was measured by releasing the cells with trypsin/EDTA and enumerating them using a particle counter (Coulter Electronics, Hialeah, FL, USA). For dose–response studies, cells were harvested on day-3. For time-course studies, cell counts were made on days 1–4.
Immunohistochemistry
Immunoperoxidase staining for E-cadherin was performed as described previously using formalin-fixed CBS cells (Sheinin et al, 2000; Chakrabarty et al, 2003). The immunoperoxidase reaction product was visualised using diaminobenzidine as the chromogenic substrate. Immunostained sections were lightly counterstained with haematoxylin and examined by light microscopy.
Preparation of cell lysates and immunoblot analysis
CBS cells were plated at 3 × 105 cells per well in wells of a six-well dish using Ca2+-free SMEM with 5% FBS as culture medium. The cells were allowed to attach overnight. The next day, they were washed and then incubated in Ca2+-free SMEM with 5% dialysed FBS with or without Ca2+ (1.4 mM). After incubation for the desired amount of time, cells were lysed in 1 × cell lysis buffer consisting of 20 mM Tris-HCl (pH 7.4), 2 mM sodium vanadate, 1.0 mM sodium fluoride, 100 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 25 μg ml−1 each aprotinin, leupeptin and pepstatin, and 2 mM EDTA and EGTA. Lysis was performed at 4°C by scraping the cells into lysis buffer and sonicating the samples. Cell lysates were incubated on ice for 15 min and then cleared by microcentrifugation at 16 000 g for 15 min. The supernatant fluids were collected and protein concentration was estimated using the BioRad DC protein assay kit (BioRad, Hercules, CA, USA).
Cell extracts containing equivalent amounts of protein (40 μg of total protein per lane) were examined by Western blotting for E-cadherin, CaSR, and signalling intermediates (total and phosphorylated forms of ERK) using procedures described previously (Bhagavathula et al, 2004). Briefly, samples were separated in SDS-PAGE under denaturing and reducing conditions and transferred to nitrocellulose membranes. After blocking with a 5% nonfat milk solution in Tris-buffered saline with 0.1% Tween (TTBS) at 4°C overnight, membranes were incubated for 1 h at room temperature with the desired antibody, diluted 1 : 1000 in 0.5% nonfat milk/0.1% TTBS. Thereafter, the membranes were washed with TTBS and bound antibody detected using the Phototope-HRP Western blot detection kit (Cell Signaling Technologies Inc.). Images were scanned, digitised and quantified using NIH image analysis software.
Statistical analysis
Data from studies with multiple experiments and multiple samples per data point were expressed as mean±s.e. Data from studies with multiple experiments and one sample per data point in each experiment (Western blot experiments) were expressed as mean±s..d. For experiments in which multiple groups were included, statistical analyses were carried out by ANOVA followed by paired group comparisons. For experiments in which two groups were compared to one another, the Student's t-test was used. P<0.05 was considered statistically significant.
Results
Effect of extracellular Ca2+ on proliferation of CBS colon cancer cells
Initially, the effects of different extracellular Ca2+ levels on proliferation of CBS cells was studied. As shown in Figure 1 (upper panel) inhibition of CBS cell proliferation was dose-dependent between 0 and 1.4 mM extracellular Ca2+. Growth was inhibited by approximately 45% in the presence of 1.4 mM Ca2+. As shown in the lower panel of Figure 1, minimal (not statistically significant) inhibition of growth was observed 1 day after shifting the cells to Ca2+ (1.4 mM)-supplemented medium. Statistically significant inhibition was observed on days 2–4. A change in the morphological appearance of the cells accompanied Ca2+-induced growth inhibition. Whereas CBS cells grew in small, grape-like clusters under low-Ca2+ conditions, they became flattened in the presence of Ca2+-supplemented medium (see Figure 2, upper panel for example).

Effect of extracellular Ca2+ on proliferation of CBS colon cancer cells. Upper panel: dose–response. Lower panel: time–response. Values shown are means and s.d. based on triplicate samples in a single experiment. Statistical significance was determined by ANOVA, followed by paired group comparisons or by Student's t-test. * indicates differences from control group (no Ca2+ supplementation) at the P<0.05 level. Both proliferation studies were conducted five times with consistent results.

Effect of U0126 on extracellular Ca2+-mediated changes in CBS cell growth and differentiation. Upper panel: morphological appearance of CBS cells grown for 3 days in Ca2+-free medium with or without 10 μ M U0126 (upper left and upper right respectively) or in medium supplemented with 1.4 mM Ca2+ with or without 10 μ M U0126 (lower left and lower right). Lower panel: CBS cell proliferation under the same conditions. Values shown are means and s.d. based on triplicate samples in a single experiment. Statistical significance was determined by ANOVA, followed by paired group comparisons. * indicates differences from high-Ca2+ group (1.4 mM Ca2+ supplementation) at the P<0.05 level. The study was conducted five times with consistent results.
Ca2+-mediated changes in CBS cell proliferation: dependence on ERK-activation
Following the observation of growth inhibition with increasing concentrations of exogenous Ca2+, we next sought to identify critical intermediary events leading from Ca2+ stimulation to reduced growth. Figure 2 shows effects of the ERK-activation inhibitor U0126 on CBS cell proliferation as well as on morphological appearance in the presence of high (1.4 mM) extracellular Ca2+. As compared to cells maintained in SMEM+dialysed FBS (without Ca2+ supplementation) throughout the 3-day incubation period, a reduction in cell growth was observed in the presence of 1.4 mM Ca2+. Concomitantly, cells maintained in Ca2+-supplemented medium underwent a change in morphological appearance (i.e. increased flattening) consistent with differentiation. Both the suppression of proliferation and induction of differentiated features were substantially reversed when U0126 (10 μ M) was included in the culture medium throughout the incubation period. These data indicate that Ca2+ modulation of growth and differentiation occurs in a MAP kinase-dependent manner.
Effects of extracellular Ca2+ and U0126 on E-cadherin expression
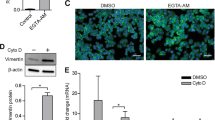
The homotypic cell–cell adhesion molecule E-cadherin functions as a tumour suppressor in colon carcinoma cells, and upregulation of E-cadherin expression is associated with the induction of differentiation in these cells (Wang and Chakrabarty, 2001; Van Aken et al, 2001). We examined the effect of extracellular Ca2+ on the expression of E-cadherin both by immunostaining and immunoblotting techniques. As shown in Figure 3 (upper panel), cells cultured in Ca2+-free SMEM plus dialysed FBS for 3 days showed little or no staining for E-cadherin. However, cells grown in the same medium supplemented with 1.4 mM Ca2+ showed significant E-cadherin expression. It could be seen that much of the reactivity in the presence of Ca2+ was at the cell periphery. When cell lysates were prepared and expression of E-cadherin assessed by Western blotting, a 3.5-fold increase in the expression of E-cadherin was observed in cells grown in Ca2+-supplemented medium as compared to the cells grown in Ca2+-free medium (Figure 3, lower panel). When U0126 (10 μ M) was included in Ca2+-supplemented medium, increased E-cadherin expression was largely abolished (Figure 3, upper and lower panels). These observations demonstrate that E-cadherin expression increases in response to extracellular Ca2+ in a MAP kinase-dependent manner.

Effect of extracellular Ca2+ and U0126 on E-cadherin expression. Upper panel: E-cadherin expression by immunostaining of CBS cells grown for three days in nonsupplemented medium (left), in medium supplemented with 1.4 mM Ca2+ (centre) and in medium supplemented with 1.4 mM Ca2+ and 10 μ M U0126 (right). Lower panel: E-cadherin expression by Western blotting (same conditions). Quantitative values are based on densitometry scanning of bands in four separate experiments and are means and s.d. Statistical significance was determined by ANOVA, followed by paired group comparisons. *indicates differences from high-Ca2+ group (1.4 mM Ca2+ supplementation) at the P<0.05 level.
In addition to using U0126 to interfere with activation through the ERK pathway, we also utilised inhibitors to block signalling through two other MAP kinase pathways (P38 and JNK) as well as signalling through the PI3K/Akt pathway. When CBS cells were treated with the P38 inhibitor, SB203580 (15 μ M), there was no effect on cell growth under low-Ca2+ conditions and only a modest (not statistically significant) increase in proliferation compared to control cells in high-Ca2+ medium (Figure 4). Consistent with this, there was an increased expression of E-cadherin in both control and SB203580-treated cells under high-Ca2+ conditions compared to cells in low-Ca2+ conditions. In contrast, there was no significant difference in E-cadherin expression between control and SB203580-treated cells (under either condition) (Figure 4). Results with the other two inhibitors were considerably different. Both the JNK inhibitor (SP600125; 0.1 μ M) and the PI3K inhibitor (LY294002; 30 μ M) strongly inhibited proliferation of CBS cells under low-Ca2+ conditions but little effect under high-Ca2+ conditions (Figure 4). Consistent with growth-inhibition under low-Ca2+ conditions was a slight increase in E-cadherin expression with both agents. Also consistent, the high level of E-cadherin expressed in Ca2+-supplemented medium was not further increased by either agent (Figure 4).

Effect of P38, JNK and PI3K inhibitors on CBS cell growth and E-cadherin expression under low-Ca2+ and high-Ca2+ conditions. Upper panel: CBS cell proliferation. Values shown are means and s.d. based on quadruplicate samples in duplicate experiments. Statistical significance was determined by ANOVA, followed by paired group comparisons. * indicates differences from the respective low-Ca2+ or high-Ca2+ control groups at the P<0.05 level. Lower panel: E-cadherin expression and β-tubulin expression by Western blotting (same conditions). Western blots were conducted in three separate experiments with consistent results.
Effects of U0126 on CaSR expression
Since the effects of extracellular Ca2+ on CBS growth and differentiation appeared to involve ERK activation, experiments were conducted to determine if the same treatment influenced CaSR expression. Consistent with our recent studies (Chakrabarty et al, 2003, 2005), CaSR was detected in CBS cells maintained for 3 days in Ca2+-free medium (SMEM+dialysed FBS), but the expression level was low. When the extracellular Ca2+ concentration was increased to 1.4 mM, expression of CaSR was substantially increased (Figure 5). Of interest, upregulation of CaSR expression did not appear to depend on ERK signalling since cells incubated in Ca2+-supplemented medium containing 10 μ M U0126 showed the same upregulation as cells maintained in the absence of the inhibitor (Figure 5).

Effect of extracellular Ca2+ and U0126 on CaSR expression. Western blotting for CaSR expression in CBS cells grown for three days in nonsupplemented medium (left), in medium supplemented with 1.4 mM Ca2+ (centre) and in medium supplemented with 1.4 mM Ca2+ and 10 μ M U0126 (right). Quantitative values are based on densitometry scanning of bands in four separate experiments and are means and s.d. Statistical significance was determined by ANOVA, followed by paired group comparisons.
Effect of extracellular Ca2+ on expression of phosphorylated-ERK
Next, CBS cells were incubated for 1 day in SMEM with dialysed FBS with or without Ca2+ and treated with U0126. Lysates were prepared from the cells at various times after treatment and probed for phosphorylated ERK and total ERK protein expression. As shown in Figure 6, there was no effect on ERK phosphorylation 5 min after exposure to extracellular Ca2+ but an increase was observed at 15 and 30 min post-treatment. ERK phosphorylation remained elevated through 1 day. When lysates prepared from cells treated with 10 μ M U0126 in Ca2+-supplemented medium were examined, ERK-phosphorylation was reduced to below basal level. Neither Ca2+-supplementation nor U0126 had a measurable effect on total ERK protein levels (Figure 6).

Effect of extracellular Ca2+ and U0126 on expression of phosphorylated-ERK. Upper panel: time-course for Ca2+-induced ERK phosphorylation. Lysates were prepared from CBS cells at various times after exposure to Ca2+ and analysed for phosphorylated-ERK by Western blotting. Lower panel: inhibition of Ca2+-induced ERK phosphorylation in the presence of U0126. CBS cells were grown for 1 day in Ca2+-free medium (left), in medium supplemented with 1.4 mM Ca2+ (centre) and in medium supplemented with 1.4 mM Ca2+ and 10 μ M U0126 (right). Lysates were prepared and analysed for ERK phosphorylation by Western blotting. Quantitative values are based on densitometry scanning of bands in four separate experiments and are means and s.d. Statistical significance was determined by ANOVA, followed by paired group comparisons. In upper panel, * indicates difference from zero-time value at P<0.05 level. In lower panel, * indicates differences from high-Ca2+ group (1.4 mM Ca2+ supplementation) at the P<0.05 level.
Differences in Ca2+ sensitivity among cloned CBS isolates
In all, 12 separate isolates of randomly-cloned CBS cells were incubated in duplicate wells. One well of each duplicate was maintained in Ca2+-free SMEM (plus dialysed FBS) while the other was incubated in the same culture medium supplemented with extracellular Ca2+ to a final concentration of 1.4 mM. After 3 days, lysates were prepared from cells in each well and examined for E-cadherin expression by Western blotting. In none of the 12 isolates was there significant E-cadherin expression in the absence of Ca2+-supplementation. In Ca2+-supplemented medium, six of the isolates expressed high levels of E-cadherin while in the other six isolates there was little or no E-cadherin expression. Western blots from representative Ca2+-responsive and -nonresponsive isolates (termed R1 and NR1, respectively) are shown in Figure 7 (upper panel). Not surprising, the isolate that expressed E-cadherin in response to Ca2+-stimulation underwent morphological differentiation in Ca2+-supplemented medium and demonstrated reduced growth as compared to cells in nonsupplemented medium (46% inhibition; n=3; P<0.05 by Student's t-test). In contrast, the cells that did not express E-cadherin in response to Ca2+-stimulation failed to differentiate (morphologically) in Ca2+-supplemented medium and did not demonstrate a significant reduction in growth (4% reduction relative to nonsupplemented medium; n=3; ns by Student's t-test).

Effect of extracellular Ca2+ on E-Cadherin, CaSR, and phosphorylated-ERK expression in Ca2+-responsive and nonresponsive CBS clones. Upper panel: E-cadherin expression; Middle panel: CaSR expression; Lower panel: ERK expression. R1 and NR1 isolates of CBS cells were grown for three days in Ca2+-free medium (left) and in medium supplemented with 1.4 mM Ca2+ (right). Western blotting was used to assess the proteins. Quantitative values are based on densitometry scanning of bands in three separate experiments and are means and s.d. Statistical significance was determined by Student's t-test, comparing nonsupplemented and Ca2+-supplemented conditions for the two isolates separately. *indicates differences from the nonsupplemented medium at the P<0.05 level.
The same Ca2+-responsive isolate R1 and Ca2+-nonresponsive isolate NR1 were maintained for 3 days in SMEM plus dialyzed FBS with or without added Ca2+. At the end of the incubation period, lysates were prepared and examined for CaSR expression by Western blotting. Both isolates expressed detectable levels of CaSR. In nonsupplemented medium, the levels expressed by the two isolates were similar to the level expressed by the uncloned parent population (Figure 7, middle panel). In Ca2+-supplemented medium, however, differences were observed. Specifically, isolate R1 showed the same upregulation of CaSR as the parent cells (compare Figure 7, middle panel with Figure 5). In contrast, isolate NR1 failed to upregulate CaSR under the same conditions (Figure 7, middle panel).
In a final set of experiments, the phosphorylation pattern of ERK was assessed in R1 and NR1 isolates grown for 1 day in SMEM plus dialysed FBS with or without added Ca2+. As shown in the lower panel of Figure 7, there was a significant increase in ERK phosphorylation in Ca2+-supplemented medium in both isolates, as compared to what was seen in nonsupplemented medium. There were no differences in the total ERK protein levels between the two isolates under either condition. Not surprisingly, U0126 (10 μ M) suppressed levels of phosphorylated ERK to below baseline levels in both isolates in Ca2+-supplemented medium (not shown).
Discussion
Extracellular Ca2+ is a critical regulator of epithelial cell growth and differentiation, but the mechanistic events that lead from an increase in extracellular Ca2+ to growth inhibition and onset of differentiation are not fully understood. Induction of E-cadherin is known to be important. E-cadherin is minimally expressed in rapidly-growing epithelial cells in vitro but is upregulated during differentiation (Birchmeier 1995; Mareel et al, 1996). In vivo, the loss of E-cadherin from the cell surface occurs during tumour progression to a more malignant phenotype (Birchmeier 1995; Mareel et al, 1996; Van Aken et al, 2001). This occurs in conjunction with a loss of differentiated histological features. In recent studies, we demonstrated that Ca2+-induced growth arrest and differentiation in CBS colon carcinoma cells were associated with E-cadherin elaboration and, ultimately, with cell surface E-cadherin expression. Among the events associated with E-cadherin accumulation in the membrane was a reduction in intra-nuclear complexes of β-catenin/TCF4 (possibly due to competition of surface E-cadherin for β-catenin) and downregulation of TCF4 transcription. Additional events associated with Ca2+ treatment in CBS cells included increased production of the cell cycle inhibitors P21 and P27, increased γ-catenin expression and increased cytokeratin expression (Chakrabarty et al, 2003, 2005; Giles et al, 2003; Kamei et al, 2003). These changes, alone or in combination, may ultimately be responsible for the growth-suppression and onset of differentiation seen in the colon epithelial cells.
The present study provides evidence that MAP kinase signalling and CaSR upregulation are both critically involved as upstream regulators of Ca2+-induced E-cadherin expression (and the subsequent changes in growth and differentiation) in cells derived from human colon cancer. The two events appear to be, at least in part, independent of one another. Using a line of colon carcinoma cells, we found that cells equilibrated in Ca2+-free medium and then exposed to 1.4 mM Ca2+ underwent a (relatively) slow but sustained rise in phosphorylated ERK. The same conditions that led to a rise in ERK phosphorylation (activation) also resulted in expression of E-cadherin and with reduced growth and increased differentiation. U0126, a potent inhibitor of MEK-dependent ERK-activation, prevented Ca2+-induced E-cadherin expression. Concomitantly, the reduction in cell proliferation and onset of differentiation normally seen in Ca2+-supplemented medium did not occur. U0126 was effective at 10 μ M, a concentration at which the inhibitor is known to be highly specific for MEK-dependent ERK activation (Favata et al, 1998). Inhibitors of three other signalling pathways did not duplicate these effects.
The ERK family of MAP kinases consists of a cascade of intracellular signalling elements that respond to a number of extracellular stimuli, including soluble peptide growth factors and components of the extracellular matrix. Peptide growth factors induce a sharp rise in activation (20–50-fold over baseline). Activation tends to be rapid (peaks within 2–5 min) and transient, with the active kinases falling to some intermediate level within 15–30 min. Under such conditions, a proliferative response is seen (Zheng et al, 1994; Chen et al, 1996; Zeigler et al, 1999). MAP kinase activation following cell contact with the extracellular matrix (integrin-mediated) results in a smaller increase (in the range of 2–6-fold increase over baseline) but the response is sustained. MAP kinase activation in this manner is associated with a differentiation response rather than with proliferation (Marshall, 1995).
Only a relatively few previous studies have addressed the relationship between MAP kinase signalling and E-cadherin expression. The data are complex and seemingly inconsistent. In one recent study, a switch from low- to high- extracellular Ca2+ was shown to induce ERK activation in an immortalised epidermal keratinocyte line (Pece and Gutkind, 2000) – similar to what we have observed here. In that study, however, E-cadherin was purposed to be a critical intermediate in the MAP kinase activation sequence since disruption of E-cadherin function with an anti-E-cadherin antibody prevented the rise in MAP kinase signalling. In another study, epithelial cells were maintained under sparse conditions (undifferentiated state) or in dense culture (where differentiation occurred) and examined for ERK expression. Cells in sparse culture expressed high levels of phosphorylated ERK relative to what was seen in dense culture. In the presence of an ERK inhibitor (PD98059), levels of phosphorylated ERK were reduced. Concomitantly, E-cadherin expression was increased (Conacci-Sorrell et al, 2003). Based on this, it was concluded that ERK activation was responsible for suppression of E-cadherin, and for preventing differentiation in sparse culture. In yet another study, MAP kinase blockade was found to suppress growth of colon tumours in vivo (Sebolt-Leopold et al, 1999). Presumably, growth suppression in vivo reflected interference with signals generated via interaction of cell surface receptors with peptide growth factors. While our data do not conflict, a priori, with these previous observations, it is clear from the present study that in the CBS colon carcinoma cells, MAP kinase activation is a critical component in the sequence of events that leads from extracellular Ca2+ stimulation to the induction of E-cadherin, and, ultimately, to growth suppression and onset of differentiation. Based on the previous findings of others (reviewed in Alpin et al, 1998), we can suggest that interaction of CBS colon carcinoma cells with components of the extracellular matrix constitutes the initial stimulus for ERK activation and the subsequent downstream events (including, presumably, E-cadherin expression) that follow. While direct evidence to support this hypothesis is lacking, what is clear is that in CBS cells, ERK activation is upstream in the sequence of events that lead from Ca2+ stimulation to growth arrest and differentiation.
Induction of CaSR appears to be a second critical event leading from Ca2+ stimulation to growth arrest and differentiation. This is based on the observation that in the mixed population of parental CBS cells, there are cells that do not respond to Ca2+ stimulation with the expected induction of E-cadherin or in growth-retardation. When these cells were isolated and cloned, they also failed to demonstrate upregulation of CaSR in response to Ca2+ stimulation. How Ca2+ stimulation leads to CaSR induction is not fully understood. The CaSR gene has promoter regions that are sensitive to both Ca2+ and vitamin D (Canaff and Hendy 2002). Response elements for the two agonists are different, such that a mutation in one site can affect binding of one agonist without affecting the other (Chakrabarty et al, 2005). Studies are underway at present to determine if clones of CBS cells that do not undergo growth arrest or differentiation in response to Ca2+ stimulation have defects in the Ca2+-binding region of the CaSR promoter. If Ca2+ is a direct activator of CaSR, one would predict (consistent with what we have observed) that downstream signalling events would not be required for induction. Additional studies will be needed to verify this suggestion and to fully explore the mechanism of Ca2+-induced CaSR expression.
The findings in the present report are based on studies in a single line of moderately-differentiated human colon carcinoma cells. However, our findings are probably not unique to these cells. In a recent study, we showed that in four different human colon carcinoma cell lines (including CBS), raising the extracellular Ca2+ concentration resulted in induction of E-cadherin and inhibition of growth (Chakrabarty et al, 2003). A direct relationship between CaSR expression and the differentiated phenotype has also been observed in both the normal colonic mucosa and malignant colonic epithelium (Sheinin et al, 2000; Chakrabarty et al, 2003, 2005).
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alpin AE, Howe A, Alahari SK, Juliano RL (1998) Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, immunoglobulin-cell adhesion molecules and selectins. Pharmacol Rev 50: 197–263
Baron JA, Beach M, Mandel JS, Van Stolk RU, Haile RW, Sandler RS, Rothstein R, Summers RW, Snover DC, Beck GJ, Bond JH, Greenberg ER (1999) Calcium supplements for the prevention of colorectal adenomas. Calcium Polyp Prevention Study Group. N Engl J Med 340: 101–107
Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Nat Acad Sci (USA) 98: 13681–13686
Bhagavathula N, Nerusu KC, Lal A, Ellis CN, Chittiboyina A, Avery MA, Ho CI, Benson SC, Pershadsingh HA, Kurtz TW, Varani J (2004) Rosiglitazone inhibits proliferation, motility, and matrix-metalloproteinase production in keratinocytes. J Invest Dermatol 122: 130–139
Birchmeier W (1995) E-cadherin as a tumor (invasion) suppressor gene. Bioessays 17: 97–99
Canaff L, Hendy GN (2002) Human calcium-sensing receptor gene, Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1, 25-dihydroxyvitamin D. J Biol Chem 277: 3037–3050
Chakrabarty S, Appelman H, Varani J (2005) Calcium sensing receptor in human colon carcinoma: Interaction with Ca2+ and 1, 25-dihydroxyvitamin D3. Cancer Res 65: 493–498
Chakrabarty S, Radjendirane V, Appelman H, Varani J (2003) Extracellular calcium and calcium sensing receptor function in human colon carcinomas: promotion of E-cadherin expression and suppression of beta-catenin/TCF activation. Cancer Res 63: 67–71
Chen Q, Lin TH, Der CJ, Juliano RL (1996) Integrin-mediated activation of MEK and mitogen-activated protein kinase is independent of Ras. J Biol Chem 271: 18122–18127
Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze'ev A (2003) Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of β-catenin signaling, Slug, and MAPK. J Cell Biol 163: 847–857
Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273: 18623–18632
Garrett JE, Capuano IV, Hammerland LG, Hung BC, Brown EM, Hebert SC, Nemeth EF, Fuller F (1995) Molecular cloning and functional expression of human parathyroid calcium receptor cDNAs. J Biol Chem 270: 12919–12925
Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 1653: 1–24
Hebert SC, Brown EM (1995) The extracellular calcium receptor. Curr Opin Cell Biol 7: 484–492
Kamei J, Toyofuku T, Hori M (2003) Negative regulation of p21 by beta-catenin/TCF signaling: a novel mechanism by which cell adhesion molecules regulate cell proliferation. Biochem Biophys Res Commun 312: 380–387
Kampman E, Slattery ML, Caan B, Potter JD (2000) Calcium, vitamin D, sunshine exposure, dairy products and colon cancer risk (United States). Cancer Causes Control 11: 459–466
Lamprecht SA, Lipkin M (2003) Chemoprevention of colon cancer by calcium, vitamin D and folate: molecular mechanisms. Nat Rev Cancer 3: 601–614
Lipkin M (1999) Preclinical and early human studies of calcium and colon cancer prevention. Ann N Y Acad Sci 889: 120–127
Mareel M, Berx G, Van Roy F, Bracke M (1996) Cadherin/catenin complex: a target for antiinvasive therapy? J Cell Biochem 61: 524–530
Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: Transient vs sustained extracellular signal-regulated kinase activation. Cell 80: 179–185
McCullough ML, Robertson AS, Rodriguez C, Jacobs EJ, Caho A, Carolyn J, Calle EE, Willett WC, Thun MJ (2003) Calcium, vitamin D, dairy products, and risk of colorectal cancer in the Cancer Prevention Study II Nutrition Cohort (United States). Cancer Causes Control 14: 1–12
Moon SE, Bhagavathula N, Varani J (2002) Keratinocyte stimulation of matrix metalloproteinase-1 production and proliferation in fibroblast: regulation through mitogen-activated protein signaling events. Br J Cancer 87: 457–464
Panka DJ, Mano T, Suhara T, Walsh K, Mier JW (2001) Phophatidylinositol 3-kinase/AKT activity regulates c-FLIP expression in tumor cells. J Biol Chem 276: 6893–6896
Pece S, Gutkind JS (2000) Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem 275: 41227–41233
Rutten MJ, Bacon KD, Marling KL, Stoney M, Meichsner CL, Lee FP, Hobson SA, Rodland KD, Sheppard BC, Trunkey DD, Deveney KE, Deveney CW (1999) Identification of Ca2+ sensing receptor in normal human gastric mucous epithelial cells. Am J Physiol 277: G662–670
Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 5: 810–816
Sellers TA, Bazyk AE, Bostick RM, Kushi LH, Olson JE, Anderson KE, Lazovich D, Folsom AR (1998) Diet and risk of colon cancer in a large prospective study of older women: an analysis stratified on family history (Iowa, United States). Cancer Causes Control 9: 357–367
Sheinin Y, Kallay E, Wrba F, Kriwanek S, Peterlik M, Cross HS (2000) Immunocytochemical localization of the extracellular calcium-sensing receptor in normal and malignant human large intestinal mucosa. J Histochem Cytochem 48: 595–602
Van Aken E, De Wever O, Correia da Rocha AS, Mareel M (2001) Defective E-cadherin/catenin complexes in human cancer. Virchows Arch 439: 725–751
Wang H, Chakrabarty S (2001) Requirement of protein kinase Cα, extracellular matrix remodeling and cell-matrix interaction for transforming growth factor β-regulated expression of E-cadherin and catenins. J Cell Physiol 187: 188–195
Wargovich MJ, Jimenez A, McKee K, Steele VE, Velasco M, Woods J, Price R, Gray K, Kelloff GJ (2000) Efficacy of potential chemopreventive agents on rat colon aberrant crypt formation and progression. Carcinogenesis 21: 1149–1155
Wu K, Willett WC, Fuchs CS, Colditz GA, Giovannucci EL (2002) Calcium intake and risk of colon cancer in women and men. J Natl Cancer Inst 94: 437–446
Zeigler M, Zeigler ME, Chi Y, Schmidt T, Varani J (1999) The role of ERK and JNK signaling pathways in growth factor-simulated cell invasion. J Cell Physiol 180: 271–284
Zheng CF, Ohmichi M, Saltiel AR, Guan KL (1994) Growth factor induced MEK activation is primarily mediated by an activator different from c-raf. Biochemistry 33: 5595–5599
Acknowledgements
This study was supported in part by Grants CA47775 and DK59169 from the National Institutes of Health, Bethesda, MD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Bhagavathula, N., Kelley, E., Reddy, M. et al. Upregulation of calcium-sensing receptor and mitogen-activated protein kinase signalling in the regulation of growth and differentiation in colon carcinoma. Br J Cancer 93, 1364–1371 (2005). https://doi.org/10.1038/sj.bjc.6602852
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602852
Keywords
This article is cited by
-
Calcium sensing receptor stimulates breast cancer cell migration via the Gβγ-AKT-mTORC2 signaling pathway
Journal of Cell Communication and Signaling (2022)
-
Epigenetic inactivation of calcium-sensing receptor in colorectal carcinogenesis
Modern Pathology (2011)
-
A Short Primer on the Calcium Sensing Receptor: An Important Cog in the Colon Cancer Wheel?
Digestive Diseases and Sciences (2011)
-
Human colon tissue in organ culture: calcium and multi-mineral-induced mucosal differentiation
In Vitro Cellular & Developmental Biology - Animal (2011)
-
Human colon tissue in organ culture: preservation of normal and neoplastic characteristics
In Vitro Cellular & Developmental Biology - Animal (2010)
