
Abstract
Recently, considerable interest has been directed to red-fluorescence photodiagnosis of brain and other tumours during surgery using the protoporphyrin IX natural precursor, 5-aminolaevulinic acid. In the present study we focused on the role of the rate-limiting enzyme porphobilinogen deaminase in glioma C6 cell activity, differentiation and sub-cellular distribution. Over-expression of the human housekeeping porphobilinogen deaminase in the glioma cells, using the housekeeping-porphobilinogen deaminase plasmid, induced a G1 cell cycle attenuation accompanied by increases in enzyme activity and c6 differentiation toward astrocytes. Visualisation of subcellular localisation of the porphobilinogen deaminase using the independent techniques of fluorescence immuno-staining with specific anti-human porphobilinogen deaminase antibodies and cellular expression of porphobilinogen deaminase fused to green fluorescent protein, revealed (unexpectedly) a major fraction of porphobilinogen deaminase in the nucleus and only a minor fraction in the cytoplasm. Both C and N terminals of porphobilinogen deaminase fused to green fluorescent protein revealed a major fraction of the newly synthesized fused porphobilinogen deaminase in the nucleus. Furthermore, newborn rat brain cells grown in a primary culture showed the same localisation pattern of porphobilinogen deaminase in the nuclei. Stimulation of C6 glioma cell differentiation by butyrate induced a marked decrease in porphobilinogen deaminase both in the nucleus and in the cytoplasm as determined by Western blotting and fluorescence immuno-localisation. These findings suggest a possible dual role for housekeeping porphobilinogen deaminase in fast dividing glioma cells, one related to the porphyrin synthesis pathway and another coupled to nuclear function, which might be linked to tumorigenesis.
Similar content being viewed by others

Main
Malignant gliomas accumulate fluorescing protoporphyrin intra-cellularly after exposure to the natural precursor 5-aminolevulinic acid (ALA), similar to a variety of tumours and leukemic cells (Malik and Lugaci, 1987; Malik et al, 1989, 1996, 1998; Peng et al, 1997a, b; Stummer et al, 1998a, b; Kennedy et al, 1990; Kennedy and Pottier, 1992). This phenomenon has been exploited for complete removal of malignant gliomas (Stummer et al, 1998a, b). The intraoperative identification of tumor tissue was performed by application of ALA prior to surgery and subsequent observation with the operating microscope with excitation using a violet-blue (375–440 nm) xenon light, a 455-nm long-pass filter and verification by analysis of fluorescence spectra. Protoporphyrin-fluorescence guided-resection resulted in residual tumour removal, which improved patient prognosis (Stummer et al, 1998a, b).
The biosynthetic pathway of protoporphyrin and heme (Kennedy et al, 1990; Peng et al, 1997a, b) consists of several enzymatic steps: (1) Glycine + Succ.CoA → 5-aminolevulinic acid (ALA) → porphobilinogen (PBG); (2) 4PBG → uroporphyrinogen → coproporphyrinogen → protoporphyrin + Fe++ → heme. All heme synthesis enzymes are exclusively localized in the cytoplasm except for the first enzyme (ALA-synthase) and the two last enzymes (protoporphyrinogen oxidase and ferrochelatase) that are known to function as mitochondrial enzymes. Jorgensen et al (2000) recently showed in paraffin embedded specimens that porphobilinogen deaminase (PBGD), the most studied enzyme in heme synthesis, is localized in the cytoplasm of rat kidney. Supplementation of ALA circumvents the rate-limiting enzyme ALA synthase (Malik and Lugaci, 1987) and enhances the accumulation of protoporphyrin if the activity of the PBGD, at the second check point, is sufficiently high. One possible rationale for the specificity of protoporphyrin accumulation in rapidly dividing cancer cells is that their enhanced metabolism demands additional heme, which is needed for increased aerobic ATP supply connected to the energy demands of the tumour. Malignant transformation of various cell lines by retrograde transforming viruses and other means leads to increased PBGD activity (Rasetti et al, 1963; Kondo et al, 1993). Similarly, the malignant lymphoproliferative diseases of chronic lymphocytic leukaemia and lymphoma were found to have higher PBGD activity in their peripheral lymphocytes than normal control subjects (Leibovici et al, 1988). However, it is unclear whether the increase in PBGD activity was caused by malignant transformation or whether it was dependent on the increased growth rate.
The purpose of the present study is to elucidate the role of PBGD in C6 glioma cells, focusing on the correlation between cell proliferation and subcellular localisation of the protein. Our findings indicate possible nuclear functions of PBGD, which may be involved in the proliferation and differentiation of glioma cells apart from the basic cytoplasmic enzymatic activity of PBGD in porphyrin synthesis.
Materials and methods
Cell culture
Rat C6 glioma cells were grown in a DMEM medium (Biological Industries, Kibbutz Beit-Haemek, Israel), supplemented with 10% foetal calf serum and antibiotics, on tissue culture plates (Corning, Staffordshire, UK) and incubated at 37°C in a humidified atmosphere with 8% CO2. The cells were recultured twice a week, using Trypsin-EDTA for detachment.
Primary newborn brain cell culture
Newborn rats brains were removed under sterile conditions, washed with PBS followed by trypsinisation under stirring for 10 min at room temperature. The supernatant was removed into a 50 cc test tube containing foetal calf serum (Biological Industries, Beit Haemek, Israel). After 10 min of centrifugation at 1200 r.p.m., the pellet was resuspended in DMEM supplemented with foetal calf serum, L-glutamine and antibiotic. The primary cell culture was plated for 20 h at the same conditions as the C6 cells.
Preparation of human PBGD cDNA expression vectors
The coding region of the erythrocyte PBGD cDNA was amplified from a human spleen cDNA library in bacteriophage λGT10 (a gift from P Sankhavarar, Yale University) using 5′-GAAGATCTATGAGAGTGATTCGCGTGGGTACC-3′ forward primer and 5′-GGAATTCTTAATGGGCATCGTTAAGCTGCCG-3′ reverse primer. The PCR product was used as a template for a PCR reaction using the forward primer: 5′-GAAGATCTATGTCTGGTAACGGCAATGCGGCTGCAACGGCGGAAGAAAACAGCCCAAAGATGAGAGTGATTCGCGTGGGT-3′ and the reverse primer 5′-GGAATTCTTAATGGGCATCGTTAAGCTGCCG-3′. The PCR product was inserted into the pLY-3 plasmid (pEGFP-C1 plasmid [Clontech, Palo Alto, CA, USA]) we created using SmaI and Eco47III restriction enzymes that lacked the green fluorescent protein (GFP) coding region, yielding a new plasmid called pHK-PBGD plasmid, which expresses the housekeeping PBGD (HK-PBGD) in mammalian cells.
The PBGD coding region was cut out from pHK-PBGD using BglII and EcoRI restriction enzymes and it was ligated to pEGFP-C1 or pEGFP-N1 (Clontech, Palo Alto, CA, USA) in frame to the C- or N-terminal of GFP. The ligation product expressed EGFP fused to HK-PBGD. Constructs were verified by sequencing (Weizmann Institute Biological Services, Rehovot, Israel).
Cell transfection with mitochondrial-GFP and PpIX localisation
For transient transfection, C6 cells were incubated with serum-free medium for 40 min followed by 6 h incubation with transfection solution containing 0.65 μg ml−1 of mitochondrial GFP plasmid (pEGFP-Mito) (Clontech, Palo Alto, CA, USA) (encoding the GFP which translocalised specifically into mitochondria) and 6.7 mg ml−1 lipofectamine reagent (GIBCO-BRL, NY, USA) in DMEM. Following transfection, the cells were transferred to a rich medium containing serum and antibiotic. Six hours before detection of PpIX localisation in the transfected cells, the cells were incubated with serum-free medium containing 0.6 mM amino-levulinic acid (Sigma, St. Louis, USA).
Stable and transient cell transfection
The pHK-PBGD plasmid was transfected into C6 cells, using lipofectamine reagent (GIBCO-BRL, NY, USA) according to the manufacturer's protocol. For stable transfection, a selective medium containing 850 μg ml−1 neomycin, also known as geneticin 418 (GIBCO-BRL, NY, USA) was added to the cells 24 h post-transfection. The surviving neomycin-resistant colonies on each plate were then subcloned by dilution and grown to establish the sub-lines. For selection maintenance, the cells were selectively grown in a medium containing 100 μg ml−1 geneticin. For PBGD fused to GFP as a transient transfection, the procedure was exactly as mentioned above, without adding neomycin. The cells were examined 24 h post transfection.
Western blotting
Proteins were quantified using the Bradford assay (Bio-Rad, CA, USA) and resolved in a 12% polyacrylamide gel. Thereafter, proteins were transferred onto nitrocellulose membranes using a semi-dry transfer apparatus (Bio-Rad, CA, USA). After blocking of the membranes with 5% skim milk and 0.06% Tween-20 in PBS, membranes were incubated with primary PBGD antibody (a generous gift from HemeBiotech, Sweden), and goat anti-rabbit secondary antibody (Jackson Immuno-Research, Pennsylvania, PA, USA) in the same solution. Immuno-reactive proteins were visualised with an enhanced chemiluminescence detection kit (Amersham, Little Chalfont, UK) used as recommended by the manufacturer.
PBGD, glial fibrilary acid protein (GFAP) and vimentin immunolabelling, and GFP–PBGD detection
Cells were seeded in eight-well slide chambers. Twenty-four hours later, cells were fixed using 4% paraformaldehyde and subsequently treated with 0.5% Triton X-100 for 30 min. Blocking was carried out with 6% skim milk, 3% BSA<and 0.2% Tween-20 in 100% FCS. Cells were then exposed to one of the next primary antibodies overnight at 4°C: Rabbit polyclonal anti-PBGD antibody (HemeBiotech, Stockholm, Sweden), rabbit polyclonal anti-GFAP antibody (Dakopatts, Glostrup, Denmark) or mouse monoclonal anti-vimentin antibody (Dakopatts, Glostrup, Denmark) Reacting rhodamine-conjugated anti-rabbit and anti-mouse antibodies (Jackson Immuno-Research, Pennsylvania, PA, USA). The cells were visualized with fluorescence microscopy (Olympus AX70).
Fluorescence microscopy
Fluorescence microscopy was performed using an Olympus AX70 microscope attached to a SpectraCube™ SD-200 spectral imaging system (Applied Spectral Imaging, Migdal HaEmek, Israel) operated as described by Malik et al (1996). Photography was carried out with an × 60 objective. For Rhodamine the excitation was at 570 nm and an image acquired at 610–700 nm; for GFP, excitation was at 420 nm and emission at 515–630 nm.
PBGD enzymatic activity assay
PBGD is assayed by determining the absorbance of uroporphyrin that is formed by light-induced oxidation of uroporphyrinogen, which is the immediate product of the enzymatic deamination. 106 C6 cells were detached from plates 24 h after plating, and resuspended in PBS without Ca2+/Mg2+. After centrifugation, the pellet was resuspended in 1 ml 50 mM tris (pH 8.2). 250 μl lysate were incubated with an equivalent amount of Triton-X-100 contained tris buffer and 100 μl of 0.5 mM porphobilinogen (PBG) (Porphyrin Products, UT, USA) for 1 h at 37°C with shaking. The reaction was stopped by addition of 10% TCA under exposure to ambient room light at room temperature for 10 min. After 10 min centrifugation (3300 r.p.m.), the supernatant was collected to the spectrofluorometer (ex. 409 nm, em. 595 nm) (Spectronic Instruments, Leeds, UK). PBGD specific activity is expressed as pmol uroporphyrin formed per mg protein per 1 h units.
Nucleus–cytoplasm fractionation
C6 cells were harvested and resuspended in Tris-HCl buffer supplemented with EDTA, EGTA and the antiproteases leupeptine, aprotinine, PMSF and DTT (Sigma, St. Louis, USA). For nucleus–cytoplasm fractionation, cells were homogenised on ice followed by centrifugation at 2500 r.p.m. for 10 min (twice), and the nuclear fraction was resuspended in the above buffer. For Western blotting of each fraction, Triton-X-100 was added and the samples were immersed in nitrogen liquid and thawed at 37°C intermittently, followed by centrifugation at 14 000 r.p.m. for 20 min.
Results
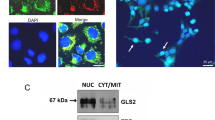
The capacity of C6 glioma cells to synthesize protoporphyrin following ALA supplementation is revealed in Figure 1; protoporphyrin accumulated in the mitochondria and cytosol. The mitochondria were identified by mitochondrial-targeted GFP (the GFP contains a mitochondrial targeting sequence directing the protein to translocalize into mitochondria), which partially co-localizes with the red protoporphyrin fluorescence.

Co-localisation of PpIX and mitochondrial-GFP in C6 glioma cells. The cells were transiently transfected with mitochondrial-GFP plasmid pEGFP-Mito and incubated for 6 h with 0.6 mM ALA. The green fluorescence of GFP and the PpIX localisation in the cells were shown by green and red filters as detected by fluorescence microscopy (left and right panels, respectively) with an × 60 objective.
In order to affirm the postulated association between PBGD activity, C6 cycle and tumour outgrowth, we over-expressed housekeeping PBGD in C6 glioma cells by stable transfection with the pHK-PBGD plasmid. Western blotting of C6 endogenous PBGD as well as of stable transfected cells over-expressing PBGD, shows an intense PBGD expression in the transfected cells (Figure 2A). As an outcome of over-expression, PBGD activity was increased fourfold from 9 to 38 pmol uroporphyrin mg−1 protein per 1 h (Figure 2B). The increased activity in PBGD was accompanied by attenuation at the G1 phase of the cell cycle as revealed by flow cytometry (Figure 3A). The G1 value of the PBGD transfected cells was 63% (Figure 3, A2) in comparison to 40% of control C6 glioma cells (Figure 3, A1) or those transfected with a control pEGFP plasmid (data not shown). The increase in G1 population of the PBGD over-expressing cells was the result of a marked reduction in the S phase fractions. No significant S/G2 transitions were revealed in this experiment. The accumulation of C6 cells in the G1 phase may represent a differentiation process leading to differentiated astrocytes. The transition from a vimentin producing phenotype of highly proliferating control cells to GFAP intermediate filament phenotype in the PBGD over-expressing cells (Figure 3B) indicates a differentiation process into astrocytes (Toda et al, 1994). An intense vimentin and basal GFAP staining was observed in non-transfected cells (Figure 3, B2 and B1, respectively), while GFAP immunofluorescence dramatically increased upon PBGD over-expression and the vimentin was down regulated (Figure 3, B3 and B4, respectively). The immunofluorescence of both vimentin and GFAP was revealed in the perinuclear region and in filametous structures, and the intense satellite-like structure of GFAP is known as typical to astrocytes.

Over-expression of PBGD in C6 glioma cells. (A) Western blotting of HK-PBGD. (B) The enzymatic activity of PBGD in control C6 and over-expressing cells (**P<0.005, as measured from five different experiments).

The effect of PBGD over-expression on the cell cycle (A) and GFAP/vimentin intermediate filaments phenotype (B) in C6 glioma cells. A1- cell cycle of C6 control cells, A2- C6 cells over-expressing the housekeeping PBGD. The GFAP/vimentin immuno-staining is shown in B. B1, B3 – immuno-staining with rabbit polyclonal anti-GFAP antibody and B2, B4 – mouse monoclonal anti-vimentin antibody immuno-staining as detected by fluorescence microscopy.
These results suggested that we attempt to visualise PBGD at the sub-cellular level. To that end we used fluorescence immuno- labelling using specific human PBGD antibodies and cellular expression of the fusion protein GFP-PBGD. These two independent techniques allowed visualisation of the subcellular localisation of endogenous as well as over-expressed PBGD in the C6 glioma cells (Figures 4 and 5). As was expected, a fraction of the PBGD immuno-fluorescence complex was found in the cytoplasm (Figure 4B,C). However, as can be seen in Figure 4B,C, the major fraction of the PBGD immuno-complex was detected in the nucleus. Nuclear PBGD localisation was a typical result in over 90% of the C6 cells in all experiments. Furthermore, newborn rat brain cells grown for 20 h as a primary culture showed the same nuclear localisation of PBGD (Figure 4D).

Nuclear localisation of PBGD in C6 cells detected by fluorescence immuno-labelling. (A) is a control in which anti-PBGD antibody was withdrawn from the samples, (B) is the endogenous PBGD, (C) is the over-expressing human housekeeping PBGD and (D) is the endogenous PBGD immuno-labelling of newborn primary brain cells. The nuclear vs cytoplasmic distribution of PBGD was a typical result in all experiments, which in most of the C6 cells was highly pronounced.

Over-expression of human housekeeping PBGD fused to GFP. Green fluorescence of C6 cells expressing GFP fused to the N-terminal (C), or to the C-terminal (D), 24 h post-transfection. The negative and positive controls for nuclear GFP localisation were cells expressing wild type GFP-N1 plasmid revealing diffused fluorescence (A) and cells expressing the SV-40 NLS GFP plasmid showing nuclear localisation (B).
Over-expression of PBGD fused to GFP (at either the C or N terminals) in the glioma cells revealed that the major fraction of the newly synthesised fused proteins is localised in the nucleus (Figure 5C,D). This confirmed the results found using immuno-labelling. Control cells transfected with wild-type GFP alone showed diffused distribution of the label throughout the cell (Figure 5A), while control NLS-GFP (Figure 5B) was localised specifically in the nucleus.
These results pointed to a possible dual role for PBGD in rapidly dividing cells, one that is related to the porphyrin synthesis pathway and another coupled to nuclear function. Therefore, the C6 glioma cells were stimulated by butyrate, to induce differentiation, and the nuclear vs cytoplasmic distribution of PBGD was analysed using Western blotting (Figure 6). In the control uninduced glioma cells, the endogenous PBGD was comparatively high both in the isolated nuclei and the cytoplasmic fraction. After 24 h of butyrate induction, PBGD decreased both in the nuclear fraction as well as in the cytoplasmic fraction. This trend was further seen after 48 h, at which time the total PBGD was reduced and it was distributed between the nucleus and cytosol. Fluorescence immuno-localisation of the PBGD in the butyrate-stimulated cells revealed a marked reduction in the nucleus and the cytoplasm (Figure 7). This result of pronounced nuclear PBGD localisation in the undifferentiated cells, and reduced nuclear and cytoplasmic distribution was found in all experiments that were carried out.

Western blot detection of PBGD localisation in C6 glioma cells during differentiation. PBGD in nuclear (upper panel) and cytoplasmic (lower panel) fractions was detected after 24 and 48 h of 2.5 mM butyric-acid treatment.

PBGD immuno-localisation in C6 glioma cells during differentiation. Intact cells are shown in the upper panel, while cells treated for 24 h with 2.5 mM butyric-acid are shown in the lower panel. This result, of pronounced nuclear PBGD localisation in undifferentiated cells and a reduced nuclear and cytoplasmic distribution, was found in all experiments that were carried out.
Discussion
The regulatory role of porphobilinogen deaminase in PpIX synthesis following ALA administration is considered the rate-limiting determinant and thus porphobilinogen deaminase is the key enzyme controlling photodiagnosis and photodynamic therapy (Gibson et al, 1998). Transient transfection of cells with the cDNA of PBGD was successful in elevating enzyme activity in both the human mammary tumour cell line MCF-7 and the human mesothelioma cell line, H-MESO-1, but this did not result in a comparable difference in the levels of PpIX (Hilf et al, 1999). PBGD has always been considered exclusively as a cytoplasmic enzyme of the heme biosynthesis pathway (Sassa, 1990). Jorgensen et al (2000) have shown immuno-histochemical localisation of PBGD in various rat tissues, in which PBGD was unevenly distributed among various cell types in a given tissue. However, nuclear localisation was not indicated. Interestingly, our present results reveal that a major fraction of the PBGD is localised specifically to the nucleus, while a relatively minor fraction is found in the cytoplasm. Since our study discusses an additional, novel, function of a known protein, which derived from its sub-cellular localisation, we confirmed the localisation findings also by immuno-labelling and GFP-tagging. The independent localisation methods, both demonstrating a nuclear fraction of PBGD, strongly indicate a nuclear-specific transport and a possible nuclear function of this protein. Nuclear localisation was seen for both the endogenous PBGD and the over-expressed human-housekeeping PBGD in glioma cells. Tagging the GFP to either the N or C termini showed the same results, which means that the PBGD nuclear localisation signal is not located in the ends of the protein.
It is well documented that nuclear localisation signals often contain lysine–arginine inner repeats separated by non-specific residues (Martelli et al, 1999). Sequence analysis of the human PBGD protein revealed RR X5 RK X17 RK repeats between residues 149 and 180, which may fulfil the requirements for a nuclear localisation signal. Nuclear localisation of PBGD is consistent with its involvement in G1 attenuation. The enzymatic activity of PBGD in the over-expressing cells was increased fourfold, and in addition, G1 cell cycle attenuation was revealed. It is well known that cells that pass the G1 phase are committed to complete cell division. Regulation of the G1 phase in a cell cycle is complex and involves many different families of proteins (Donjerkovic and Scott, 2000), some of which are cell-type specific and thus related to cell differentiation (Coffman and Studzinski, 1999). Our model system, based on a rapidly proliferating rat C6 glioma cell line which has oligodendrocytic, astrocytic, and neuronal properties (Parker et al, 1980; Segovia et al, 1994), is widely used to study differentiation processes. Astrocyte-like differentiation involves a shift from vimentin synthesis toward glial fibrillary acidic protein (GFAP) production (Roymans et al, 2000) followed by morphological changes. Our present results show typical morphological changes to an astrocyte-like phenotype revealing perinuclear GFAP satellite-like formation in the PBGD transfected cells. This result correlates well with a reduction in vimentin synthesis. Since these findings are joined both to G1 attenuation and to C6 differentiation in PBGD-transfected cells, we assume that PBGD plays an additional role in cell differentiation or in other cellular processes. Piatigorsky provided evidence for multiple protein functions for a single protein involved in different cellular mechanisms (Piatigorsky, 1992). The δ-aminolevulinic acid dehydratase, which is also an enzyme that participates in the heme biosynthesis pathway, has been shown to play an additional role as inhibitor of the 26S proteasome (Guo et al, 1994).
Schoenfeld showed that increased activity of PBGD is strongly correlated with an increased growth rate in both non-malignant and malignant cell lines (Schoenfeld et al, 1988). The power of elevated activity of PBGD to produce PpIX was clinically demonstrated for malignant gliomas (Stummer et al, 1998b); oral cavity cancer (Leunig et al, 2000a); colonic and gastrointestinal dysplasia (Stepp et al, 1998); peritoneal endometriosis (Hillemanns et al, 2000); laryngeal neoplasms (Leunig et al, 2000b); malignant lesions of the oesophagus (Hinnen et al, 1998); lower urinary tract tumours (Kriegmair et al, 1999); bladder cancer (Kriegmair et al, 1996); malignant mucosa in head and neck cancer (Betz et al, 1999); Barrett's oesophagus and adenocarcinoma (Stepp et al, 1998); basal and squamous cell carcinoma (Orenstein et al, 1995, 1996, 1997; Malik et al, 1998) and coetaneous lymphoma (Orenstein et al, 2000).
A direct correlation between the intracellular accumulation of PpIX and malignancy could not always be found, as in the case of Barrett's esophagus and adenocarcinoma. However, higher heme biosynthetic enzyme activities (i.e. PBGD) and lower PpIX precursor concentrations were found in squamous carcinoma (Hinnen et al, 2000).
Consistent with the increased activity of PBGD in proliferating cells, our results show decreased nuclear and cytoplasmic PBGD levels in differentiating cells. Additionally, Lindberg showed recently that mutant mice lacking the PBGD gene exhibit a marked decrease in large-caliber axons and manifest general motor neuropathy (Lindberg et al, 1999). This reduced activity of PBGD correlates with an additional role for PBGD in differentiation.
Photodiagnosis and protoporphyrin IX-dependent phototherapy are highly successful techniques with a wide range of clinical therapeutic application (Peng et al, 1997a, b). On the basis of the present findings, we may conclude that the intracellular changes in PBGD levels is intimately connected to differentiation. The markedly increased capacity of tumours to produce and accumulate PpIX, which is directly associated with elevated PBGD activity, possibly reflects an intrinsic virtue of the tumour metabolic capacity.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Betz, CS, Mehlmann, M, Rick, K, Stepp, H, Grevers, G, Baumgartner, R & Leunig, A (1999). Autofluorescence imaging and spectroscopy of normal and malignant mucosa in patients with head and neck cancer. Lasers Surg Med, 25: 323–334.
Coffman, FD & Studzinski, GP (1999). Differentiation-related mechanisms which suppress DNA replication. Exp Cell Res, 248: 58–73.
Donjerkovic, D & Scott, DW (2000). Regulation of the G1 phase of the mammalian cell cycle. Cell Res, 10: 1–16.
Gibson, SL, Cupriks, DJ, Havens, JJ, Nguyen, ML & Hilf, R (1998). A regulatory role for porphobilinogen deaminase (PBGD) in delta-aminolaevulinic acid (delta-ALA)-induced photosensitization? Br J Cancer, 77: 235–243.
Guo, GG, Gu, M & Etlinger, JD (1994). 240-kDa proteasome inhibitor (CF-2) is identical to delta-aminolevulinic acid dehydratase. J Biol Chem, 269: 12399–12402.
Hilf, R, Havens, JJ & Gibson, SL (1999). Effect of delta-aminolevulinic acid on protoporphyrin IX accumulation in tumor cells transfected with plasmids containing porphobilinogen deaminase DNA. Photochem Photobiol, 70: 334–340.
Hillemanns, P, Weingandt, H, Stepp, H, Baumgartner, R, Xiang, W & Korell, M (2000). Assessment of 5-aminolevulinic acid-induced porphyrin fluorescence in patients with peritoneal endometriosis. Am J Obstet Gynecol, 183: 52–57.
Hinnen, P, de Rooij, FW, van Velthuysen, ML, Edixhoven, A, van Hillegersberg, R, Tilanus, HW, Wilson, JH & Siersema, PD (1998). Biochemical basis of 5-aminolaevulinic acid-induced protoporphyrin IX accumulation: a study in patients with (pre)malignant lesions of the oesophagus. Br J Cancer, 78: 679–682.
Hinnen, P, de Rooij, FW, Terlouw, EM, Edixhoven, A, van Dekken, H, van Hillegersberg, R, Tilanus, HW, Wilson, JH & Siersema, PD (2000). Porphyrin biosynthesis in human Barrett's oesophagus and adenocarcinoma after ingestion of 5-aminolaevulinic acid. Br J Cancer, 83: 539–543.
Jorgensen, PE, Erlandsen, EJ, Poulsen, SS, Markussen, S, Koch, C & Brock, A (2000). Activity and immunohistochemical localization of porphobilinogen deaminase in rat tissues. Scand J Clin Lab Invest, 60: 635–642.
Kennedy, JC & Pottier, RH (1992). Endogenous protoporphyrin IX, a clinically useful photosensitizer for 5-Aminolevulinic acid-based photodynamic therapy: principles and experimental research. Photochem Photobiol, 65: 235–251.
Kennedy, JC, Pottier, RH & Pross, DC (1990). Photodynamic therapy with endogenous protoporphyrin IX: basic principles and present clinical experience. J Photochem Photobiol B:Biol, 6: 143–148.
Kondo, M, Hirota, N, Takaoda, T & Kajiware, M (1993). Heme-biosynthetic enzyme activities and porphyrin accumulation in normal liver and hepatoma cell line of rats. Cell Biol Toxicol, 9: 95–105.
Kriegmair, M, Baumgartner, R, Knüchel, R, Stepp, H, Hofstadter, F & Hofstetter, A (1996). Detection of early bladder cancer by 5-aminolevulinic acid induced porphyrin fluorescence. J Urol, 155: 105–110.
Kriegmair, M, Zaak, D, Knuechel, R, Baumgartner, R & Hofstetter, A (1999). 5-Aminolevulinic acid-induced fluorescence endoscopy for the detection of lower urinary tract tumors. Urol Int, 63: 27–31.
Leibovici, L, Schoenfeld, N, Yehoshua, HA, Mamet, R, Rakowski, E, Shindel, A & Atsmon, A (1988). Activity of porphobilinogen deaminase in peripheral blood mononuclear cells of patients with metastatic cancer. Cancer, 62: 2297–2300.
Leunig, A, Betz, CS, Baumgartner, R, Grevers, G & Issing, WJ (2000a). Initial experience in the treatment of oral leukoplakia with high-dose vitamin A and follow-up 5-aminolevulinic acid induced protoporphyrin IX fluorescence. Eur Arch Otorhinolaryngol, 257: 327–331.
Leunig, A, Betz, CS, Mehlmann, M, Stepp, H, Arbogast, S, Grevers, G & Baumgartner, R (2000b). A pilot series demonstrating fluorescence staining of laryngeal papilloma using 5-aminolevulinic acid. Laryngoscope, 10: 1783–1785.
Lindberg, RL, Martini, R, Baumgartner, R, Erne, B, Borg, J, Zielasek, J, Ricker, K, Steck, A, Toyka, KV & Meyer, UA (1999). Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest, 103: 1127–1134.
Malik, Z, Kostenich, G, Rothmann, C, Barshack, I & Orenstein, A (1998). Imaging of human skin lesions using multipixel Fourier transform spectroscopy. Lasers Med Sci, 13: 12–118.
Malik, Z, Dishi, M & Garini, Y (1996). Fourier transform multipixel spectroscopy and spectral imaging of protoporphyrin and Photofrin in single melanoma cells. Photochem Photobiol, 63: 608–614.
Malik, Z, Ehrenberg, B & Faraggi, A (1989). Inactivation of erythropoietic, lymphopoietic and myelocytic leukemic cells by photoexcitation of endogenous porphyrins. J Photobiol Photochem B Biol, 4: 195–205.
Malik, Z & Lugaci, H (1987). Destruction of erythroleukemic cells by photoactivation of endogenous porphyrins. Br J Cancer, 56: 589–595.
Martelli, AM, Sang, N, Borgatti, P, Capitani, S & Neri, LM (1999). Multiple biological responses activated by nuclear protein kinase. C J Biol Chem, 74: 499–521.
Orenstein, A, Haik, J, Tamir, J, Winkler, E, Trau, H, Malik, Z & Kostenich, G (2000). Photodynamic therapy of cutaneous lymphoma using 5-aminolevulinic acid topical application. Dermatol Surg, 26: 765–770.
Orenstein, A, Kostenich, G & Malik, Z (1997). The kinetics of protoporphyrin fluorescence during ALA-PDT in human malignant skin tumors. Cancer Lett, 120: 229–234.
Orenstein, A, Kostenich, G, Kogan, L & Malik, Z (1995). Temperature monitoring during photodynamic therapy of skin tumors with topical 5-aminolevulinic acid application. Cancer Lett, 93: 227–232.
Orenstein, A, Kostenich, G, Tsur, H, Katanick, D, Kopolovic, J, Ehrenberg, B, Roitman, L & Malik, Z (1996). Photodynamic therapy of malignant lesions of the skin mediated by topical application of 5-aminolevulinic acid in combination with DMSO and EDTA. Lasers Life Sci, 7: 1–9.
Parker, KP, Norenberg, MD & Vernadakis, A (1980). “Transdifferentiation” of C6 glial cells in culture. Science, 208: 179–181.
Peng, Q, Warloe, T, Berg, K, Moan, J, Kongshaug, M, Giercksky, KE & Nesland, JM (1997a). 5-Aminolevulinic acid-based photodynamic therapy. Clinical photodynamic therapy. J Photochem Photobiol B Biol, 14: 275–292.
Peng, Q, Warloe, T, Berg, K, Moan, J, Kongshaug, M, Giercksky, KE & Nesland, JM (1997b). 5-Aminolevulinic acid-based photodynamic therapy. Clinical research and future challenges. Cancer, 79: 2282–2308.
Piatigorsky, J (1992). Lens crystallins. Innovation associated with changes in gene regulation. J Biol Chem, 267: 4277–4280.
Rasetti, L, Rubino, GF, Tettinatti, L & Drago, GW (1963). Porphyrin porphobilinogen and amino ketone levels in tumor tissue. Panminerva Med, 7: 105–110.
Roymans, D, Willems, R, Vissenberg, K, De Jonghe, C, Grobben, B, Claes, P, Lascu, I, Van Bockstaele, D, Verbelen, JP, Van Broeckhoven, C & Slegers, H (2000). Nucleoside diphosphate kinase (Nm23-R1/NDPK) is associated with intermediate filaments and becomes upregulated upon camp induced differentiation of rat C6 glioma. Exp Cell Res, 261: 127–138.
Sassa, S (1990). Regulation of the genes for heme pathway enzymes in erythroid and in non-erythroid cells. Int J Cell Cloning, 8: 10–26.
Schoenfeld, N, Mamet, R, Leibovici, L, Epstein, O, Teitz, Y & Atsmon, A (1988). Growth rate determines activity of porphobilinogen deaminase both in nonmalignant and malignant cell lines. Biochem Med Metab Biol, 40: 213–217.
Segovia, J, Lawless, JM, Tillakaratne, NJK, Brenner, M & Tobin, A (1994). Cylic AMP decreases the expression of a neuronal marker (GAD 67) and increases the expression of an astroglial marker (GFAP) in C6 cells. J Neurochem, 63: 1218–1225.
Stepp, H, Sroka, R & Baumgartner, R (1998). Fluorescence endoscopy of gastrointestinal diseases: basic principles, techniques, and clinical experience. Endoscopy, 30: 379–386.
Stummer, W, Stepp, H, Moller, G, Ehrhardt, A, Leonhard, M & Reulen, HJ (1998a). Technical principles for protoporphyrin-IX-fluorescence guided microsurgical resection of malignant glioma tissue. Acta Neurochir, 140: 995–1000.
Stummer, W, Stocker, S, Wagner, S, Stepp, H, Fritsch, C, Goetz, C, Goetz, AE, Kiefmann, R & Reulen, HJ (1998b). Intraoperative detection of malignant gliomas by 5-aminolevulinic acid induced porphyrin fluorescence. Neurosurgery, 42: 518–526.
Toda, M, Miura, M, Asou, H, Toya, S & Uyemura, K (1994). Cell growth suppression of astrocytoma C6 cells by glial fibrillary acidic protein cDNA transfection. J Neurochem, 63: 1975–1978.
Acknowledgements
We thank Hemebiotech, Denmark for the generous gift of anti-human PBGD antibodies. We are grateful to Ms Judith Hanania for her skilful help during the execution of this study and Professor N Schoenfeld, Dr R Mamet and R Mevasser of the Porphyria Reference Laboratory, The Rabin Center, Israel for their assistance. This study was supported by a GIF grant no. 052-202.08/98.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Greenbaum, L., Gozlan, Y., Schwartz, D. et al. Nuclear distribution of porphobilinogen deaminase (PBGD) in glioma cells: a regulatory role in cancer transformation?. Br J Cancer 86, 1006–1011 (2002). https://doi.org/10.1038/sj.bjc.6600173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6600173
