
Abstract
Background
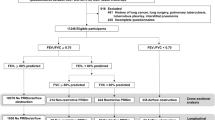
Physiologic detection of bronchiolar obstruction in children with cystic fibrosis (CF) may be clinically unsuspected because of normal routine spirometry despite bronchiectasis on lung CT.
Methods
Children from two accredited CF facilities had spirometry obtained every 3 months when clinically stable. Pre-bronchodilator maximum expiratory flow volume curves were retrospectively analyzed over 16 years to detect an isolated abnormal FEF75%, despite normal routine spirometry.
Results
At Miller Children’s and Women’s Hospital (MCWH), an abnormal FEF75% was initially detected in 26 CF children at age 7.5 ± 4 (SD) years despite normal routine spirometry initially. FEF75% remained an isolated abnormality for 2.5 ± 1.5 years after it was initially detected in these 26 CF children. At Cohen Children’s Medical Center (CCMC), despite normal routine spirometry initially, abnormal FEF75% occurred in 13 children at age 11.7 ± 4.5 years, and abnormal FEF25–75% in 10 children at age 11.8 ± 5.3 years.
Conclusions
FEF75% was most sensitive spirometric test for diagnosing both early and isolated progressive bronchiolar obstruction. Data from CCMC in older children demonstrated the simultaneous detection of abnormal FEF75% and FEF25–75% values consistent with greater bronchiolar obstruction when serial spirometry was initiated at an older age.
Impact
-
There is very little published spirometric data regarding diagnosis of isolated small airways obstruction in CF children.
-
FEF75% can easily detect unsuspected small airways obstruction in CF children with normal routine spirometry and bronchiectasis on lung CT and optimize targeted modulatory therapies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 14 print issues and online access
$259.00 per year
only $18.50 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
O’Sullivan, B. P. & Freedman, S. D. Cystic fibrosis. Lancet 373, 1891–1904 (2009).
Middleton, P. G. et al. Elexacaftor-Tezacaftor-Ivacaftor for cystic fibrosis with a single Phe508del Allele. N. Engl. J. Med. 381, 809–819 (2019).
Zemanick, E. T. et al. A Phase 3 open-label study of Elexacaftor/Tezacaftor/Ivacaftor in children 6 through 11 years of age with cystic fibrosis and at least one F508 del- CFTR Allele. Am. J. Respir. Crit. Care Med. 203, 1522–1532 (2021).
Schluchter, M. D., Konstan, M. W. & Davis, P. B. Jointly modeling the relationship between survival and pulmonary function in cystic fibrosis patients. Stat. Med. 21, 1271–1287 (2002).
Tiddens, H. A. W. M., Donaldson, S. H., Rosenfeld, M. & Pare, P. D. Cystic fibrosis lung disease starts in the small airways: can we treat it more effectively? Pediatr. Pulmonol. 45, 107–111 (2010).
Oppenheimer, E. H. & Esterly, J. R. Pathology of cystic fibrosis. Review of the literature and comparison with 146 autopsied cases. Perspect. Pediatr. Pathol. 2, 241–278 (1975).
Sobonya, R. E. & Taussig, L. M. Quantitative aspects of lung pathology in cystic fibrosis. Am. Rev. Respir. Dis. 134, 290–295 (1986).
Tiddens, H. A. W. M. et al. Cartilaginous airway wall dimensions and airway resistance in cystic fibrosis lungs. Eur. Respir. J. 15, 735–742 (2000).
Hamutcu, R. et al. Clinical findings and lung pathology in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 165, 1172–1175 (2002).
Quanjer, P. H. & Weiner, D. J. Interpretative consequences of adopting the Global Lungs 2012 Reference Equations for spirometry for children and adolescents. Pediatr. Pulmonol. 49, 118–125 (2014).
Graham, B. L. et al. Standardization of spirometry 2019 update. American Thoracic Society and European Respiratory Society Technical Statement. Am. J. Respir. Crit. Care Med. 200, e70–e88 (2019).
Goris, M. L., Zhu, H. J., Blankenberg, F., Chan, F. & Robinson, T. E. An automated approach to quantitative air trapping measurements in mild cystic fibrosis. Chest 123, 1655–1663 (2003).
Ranganathan, S. C. et al. The evolution of airway function in early childhood following diagnosis of cystic fibrosis. Am. J. Respir. Crit. Care Med. 169, 928–933 (2004).
Linnane, B. M. et al. Lung function in infants with cystic fibrosis diagnosed by newborn screening. Am. J. Respir. Crit. Care Med. 178, 1238–1244 (2008).
Bakker, E. M. et al. Small airway involvement in cystic fibrosis lung disease: routine spirometry is an early and sensitive marker. Pediatr. Pulmonol. 48, 1081–1088 (2013).
Hiatt, P., Eigen, H., Yu, P. & Tepper, R. S. Bronchodilator responsiveness in infants and young children with cystic fibrosis. Am. Rev. Respir. Dis. 137, 119–122 (1988).
Tiddens, H. A. W. M. Detecting early structural lung damage in cystic fibrosis. Pediatr. Pulmonol. Suppl. 34, 228–231 (2002).
Zapletal, A., Desmond, K. J., Demizio, D. & Coates, A. L. Lung recoil and the determination of airflow abnormalities in cystic fibrosis and asthma. Physiopathol. Respir. 15, 575–592 (1979).
Lukic, K. Z. & Coates, A. L. Does the FEF25-75 or the FEF75 have any value in assessing lung disease in children with cystic fibrosis or asthma? Pediatr. Pulmonol. 50, 863–868 (2015).
Fretzayas, A., Douros, K., Moustaki, M. & Loukou, I. Applications of lung clearance index in monitoring children with cystic fibrosis. World J. Clin. Pediatr. 8, 15–22 (2019).
de Jong, P. A. et al. Progression of lung disease on computed tomography and pulmonary function tests in children and adults with cystic fibrosis. Thorax 61, 80–85 (2006).
Stanojevic, S. et al. Determinants of lung disease progression measured by lung clearance index in children with cystic fibrosis. Eur. Respir. J. 58, 2003380 (2021).
Frauchiger, B. et al. Longitudinal course of clinical lung clearance index in children with cystic fibrosis. Eur. Respir. J. 58, 2002686 (2021).
Murphy, K. P., Maher, M. M. & O’Conner, O. J. Imaging of cystic fibrosis and pediatric bronchiectasis. Am. J. Roentgenol. 206, 448–454 (2016).
Gustatsson, P. M., DeJong, P. A., Tiddens, H. A. W. M. & Lindblad, A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 63, 129–134 (2008).
Martinez, T. M. et al. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 172, 1133–1138 (2005).
Desai, U. & Josh, J. M. Impulse oscillometry. Adv. Respir. Med. 87, 235–238 (2019).
Gelb, A. F., Gold, W. M., Wright, R. R., Bruch, H. R. & Nadel, J. A. Physiologic diagnosis of subclinical emphysema. Am. Rev. Respir. Dis. 107, 50–63 (1973).
Gelb, A. F. et al. Normal routine spirometry can mask COPD/Emphysema in symptomatic smokers. Chronic Obstr. Pulm. Dis. 8, 124–134 (2021).
Gelb, A. F. & Zamel, N. Simplified diagnosis of small-airway obstruction. N. Engl. J. Med. 288, 395–398 (1973).
Gelb, A. F. et al. Further studies of unsuspected emphysema in nonsmoking patients with asthma with persistent expiratory airflow obstruction. Chest 153, 618–629 (2018).
Egan, M. E. Cystic fibrosis transmembrane conductor receptor modulator therapy in cystic fibrosis. Curr. Opin. Pediatr. 32, 384–388 (2020).
Mayer-Hamblett, N. et al. Building global development strategies for CF therapeutics during a transitional CFTR modulator era. J. Cyst. Fibros. 19, 677–687 (2020).
O’Conner, J. B. et al. Divergence of bacterial communities in the lower airways of CF patients in early childhood. PLoS ONE 16, e0257838 (2021).
Nichols, D. P. et al. PROMISE: Working with the CF community to understand emerging clinical and research needs for those treated with highly effective CFTR modulator therapy. J. Cyst. Fibros. 20, 205–212 (2021).
Author information
Authors and Affiliations
Contributions
E.N., A.F.G., and J.A.N. contributed to conception and design of the study. V.K.M., E.N., A.F.G., Y.F., J.K.D.C.-G., and D.M. contributed to acquisition of the data. V.K.M., E.N., A.F.G., Y.F., J.K.D.C.-G., D.M., and D.S. contributed to analysis and interpretation of data. V.K.M., E.N., A.F.G, and A.F.G. drafted the article, and all authors have revised it critically for important intellectual content. All authors have approved the final version to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by Miller Children’s and Women’s Hospital, Long Beach Memorial Hospital, Long Beach, California, MHS IRB Number 843-18, and Cohen Children’s Medical Center, Northwell Health, Lake Success, New York, IRB Number 20-0581 and Clinical Trials.gov NCT 03839992. Parents/guardians of patients consented to inclusion of data in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Masson, V.K., Nussbaum, E., Gelb, A.F. et al. Isolated abnormal FEF75% detects unsuspected bronchiolar obstruction in CF children. Pediatr Res 94, 1051–1056 (2023). https://doi.org/10.1038/s41390-023-02532-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-023-02532-2
