
Abstract
Autophagy, a process for the degradation of protein aggregates and dysfunctional organelles, is required for cellular homeostasis and cell survival in response to stress and is implicated in endogenous protection. Ischemic preconditioning is a brief and nonlethal episode of ischemia, confers protection against subsequent ischemia-reperfusion through the up-regulation of endogenous protective mechanisms. Emerging evidence shows that autophagy is associated with the protective effect of ischemic preconditioning. This review summarizes recent progress in research on the functions and regulations of the autophagy pathway in preconditioning-induced protection and cellular survival.
Similar content being viewed by others


Introduction
Autophagy is an intracellular catabolic process in which the cytoplasmic constituents, such as aggregated proteins and dysfunctional organelles, are surrounded by a double membrane, termed the autophagosome, and are transported to lysosomes for degradation and recycling1,2. Autophagy plays a critical role in cellular development, differentiation and life-span extension2. Under physiological conditions, the incidence of autophagy is maintained at a low level. However, during various stress conditions, such as oxidative stress, nutrient limitation, hypoxia and endoplasmic reticulum stress, autophagy is induced and is associated with the pathological process of many diseases2,3 including cancer, neurodegenerative disorders and ischemic injury. The excessive activation of autophagy can destroy normal proteins and organelles, leading to cell death. This form of cell death is generally termed type-II programmed cell death. However, autophagy also represents an adaptive strategy of the cells. The modest activation of autophagy promotes cell survival. For example, autophagy is activated rapidly during starvation and is responsible for the turnover of unnecessary cytoplasmic proteins and damaged organelles. In this scenario, amino acids and fatty acids are extracted and recycled for the production of adenosine triphosphate (ATP)2.
Ischemic preconditioning (IPC), which is a brief and non-lethal episode of ischemia, confers protection against subsequent ischemia-reperfusion (I/R) through the up-regulation of endogenous protective mechanisms. The IPC-induced protective effect was first identified in the heart by Murry et al4 and was subsequently found in other organs, such as the brain, spinal cord and kidney5,6. Recent studies have reported that the activation of autophagy is associated with IPC and hyperbaric oxygen-induced ischemic tolerance to I/R injury7,8. In this review, we will discuss the functions and mechanisms of autophagy in the protection induced by preconditioning.
The process of autophagy
Autophagy is highly conserved from yeast to mammals. Depending on the pathways used for the delivery of intracellular cargo to lysosomes, there are three major types of autophagy, macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy. Macroautophagy sequesters cytosolic components into double membrane-formed autophagosomes, which fuse with the lysosome for degradation9,10. This degradative process is relatively nonselective and involves the formation, maturation and degradation of autophagosomes. In contrast, chaperone-mediated autophagy is a highly selective degradation pathway that degrades proteins containing the KFERQ-like peptide motif through the lysosomes11. In contrast to macroautophagy and CMA, microautophagy involves the direct engulfment of small amounts of cytoplasm by invagination, protrusion, and the separation of the lysosomal membrane12, and the process is presumed to depend upon the morphological characteristics of the lysosomes.
Autophagy is divided into four key degradation steps that begin with the formation of the phagophore. The source of the phagophore membrane is likely the endoplasmic reticulum (ER), mitochondria, Golgi complex or plasma membrane13,14. During phagophore formation, specific proteins, including Atg proteins, are recruited into the forming autophagosomal membranes. This process is regulated by two major functional groups, the Atg9 group and cycling system including Atg9, Atg2, Atg18, and the Atg1 kinase complex (Atg1 and Atg13)15, and the functional group consisting of the class III phosphatidylinositol 3-kinase (PI3K) (Vps34), Beclin 1 (yeast Atg6) and p150 (mammalian Vps15)16. The formation of autophagosomes is the second step in the autophagy process and is initiated by the emergence of the phagophore. The phagophore membranes are extended, and they surround the cytosolic constituents to form a double membrane structure termed the autophagosome. The two ubiquitin-like conjugation systems essential for this process are the Atg12-Atg5 system and the microtubule-associated protein 1 light chain 3 (LC3, yeast Atg8)-phosphatidylethanolamine (LC3-PE) system. The Atg12-Atg5-Atg16 complex is localized to the outer membrane of the extending phagophore and is required for the curvature of the extending phagophore17,18. However, the completion of the autophagosome requires the LC3-PE system. LC3-PE (LC3 II) is recruited specifically to the autophagosomal membrane, is an essential component of the autophagosome18 and is used as an autophagic marker. Following the completion of autophagosome formation, the autophagosomes are transported to the lysosomes along microtubules that depend on the dynein-dynactin complex19; therefore, the cytoskeleton plays a role in the maturation of autophagosomes20. The outer membrane of the autophagosome fuses with the lysosome to form the autophagolysosome. During this process, non-Atg components, such as ESCRT, SNAREs, Rab7, and LAMP-2, are required for the fusion of the autophagosome with the lysosome21. In the final step, the contents of the autolysosome are degraded by lysosomal hydrolases, such as lipases and cathepsins; therefore, lysosome activity is essential for functional autophagy. For example, the maturation of the autolysosome requires cathepsin proteases B and D22.
Several stress conditions, such as starvation and oxygen deprivation, induce autophagy, which is involved in the protection of organs. Increasing evidence indicates that IPC induces ischemic tolerance to I/R injury through the up-regulation of the endogenous protective mechanism, which is associated with the maintenance of ATP production and intracellular homeostasis, and inhibits apoptotic cell death. These data suggest that IPC and autophagy share many common features, and autophagic pathways play an important role in the protective effect of IPC in I/R injury.
Autophagy pathway mediates cell survival during ischemic preconditioning
AMPK and mTOR
The mammalian target of mTOR is the upstream negative regulator of autophagy in mammalian cells, especially in the absence of nutrients and energy23. During ischemic injury in the heart, the cellular ATP content decreases and AMP accumulates because of energy depletion, which activates the AMP-activated protein kinase (AMPK). The activation of AMPK suppresses mTOR activity, thereby up-regulating autophagy24, indicating that the pathway for AMPK–mTOR is important for autophagy regulation. It has been reported that in cultured cardiomyocytes exposed to glucose deprivation, the level of ATP was reduced significantly, leading to the activation of AMPK and suppression of mTOR, which was coincident with the up-regulation of autophagy. The activation of autophagy induced by myocardial ischemia was inhibited in transgenic mice overexpressing a dominant negative AMPK25. It has also been reported that the absence of AMPK aggravated myocardial infarct size in mouse hearts subjected to ischemia, which is associated with autophagosomal formation. These results suggest that the up-regulatiion of autophagy mediated by the AMPK-mTOR pathway has a beneficial effect on cell survival during ischemia24. AMPK has been shown to contribute to the protective effects of ischemic preconditioning in neurons. Therefore, it is conceivable that autophagy is involved in protection by ischemic preconditioning through the AMPK–mTOR pathway.
Recent studies have demonstrated that AMPK increases the activation of autophagy through the direct activation of Atg proteins, such as Ulk1, rather than through mTOR inhibition26. In addition, there is evidence that p53 suppresses mTOR and subsequently induces autophagy27.
Beclin 1
Beclin1, a Bcl-2 interacting protein, is an upstream regulator of autophagy in mammalian cells. In the rat brain, the up-regulation of Beclin1 inhibits the replication of the Sindbis virus and exerts a protective effect on neurons through inhibiting apoptosis28, indicating that the activation of autophagy is beneficial to neuron survival. In general, it is accepted that during ischemic injury, autophagy is induced and the expression of Beclin1 is increased. It has been demonstrated that the overexpression of Beclin1 in cultured HL-1 cells protects the cells from ischemic injury. However, the protective effect of Beclin1 was reversed by the expression of a dominant negative mutant of Atg5. Additionally, both down-regulating Beclin1 and activating the Beclin1 domain that binds to Bcl-2 reduced autophagy and increased apoptosis in cardiomyocytes29. These results suggest that the activation of autophagy plays a protective role in cardiomyocytes. Meanwhile, Beclin1 up-regulates autophagy through the direct interaction with Bcl-2, and this has been further demonstrated by Carloni et al in hypoxia–ischemia model of the neonatal brain. The study demonstrated that pretreatment with rapamycin, an activator of autophagy, increased the expression of Beclin1, thereby reducing cerebral ischemia injury and necrotic cell death30.
Despite these compelling data, there are controversies concerning the relationship between autophagy and ischemic insult, and there is conflicting evidence that Beclin1-induced autophagy leads to cell death. When the autophagy mediated by Beclin1 was inhibited through the siRNA-mediated down-regulation of Beclin1 expression in cultured cardiomyocytes, the cellular survival increased after ischemic damage31. Mice containing a heterozygous disruption of Beclin1 exhibited an inhibition of autophagy, a decrease in apoptosis and a reduction in infarct size during myocardial ischemia-reperfusion24. However, it is unknown whether the autophagy induced by Beclin1 is detrimental or protective.
PI3K
The class I phosphatidylinositol 3-kinase (PI3K) and class-III PI3K play distinct roles in autophagy. Class-I PI3K inhibits autophagy, whereas class-III PI3K induces autophagy32. For example, 3-meth-yladenine (3-MA), an inhibitor of class-III PI3K, suppresses autophagy in mammalian cells33. The inhibitory effect of class-I PI3K on autophagy is mediated through the activation of protein kinase B (Akt) following the activation of mTOR34. However, the class-III PI3K forms a complex with Beclin1 and other proteins, which recruits constituent proteins to the autophagosomal membrane, inducing autophagy35. It has been shown that during glucose deprivation in H9c2 cells, the expression of class-I PI3K increased, promoting cardiomyocyte death. Conversely, the treatment with 3-MA and leupeptin, inhibitors of autophagy, inhibited autophagy and reduced cell viability36. There are data demonstrating that rapamycin improves the survival of isolated adult cardiomyocytes exposed to glucose deprivation37. These findings indicate that autophagy enhances cell viability under conditions of energy depletion. In our previous study, we demonstrated that in the focal cerebral ischemia model, rapamycin protects the rat brain from ischemic injury. Furthermore, pretreatment with 3-MA exacerbates the cerebral injury caused by ischemia-reperfusion, suggesting that autophagy-induced neuroprotection against cerebral ischemia may be mediated by class-III PI3K8. It has been reported that the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia is likely associated with the up-regulation of autophagy and activation of the PI3K-Akt-mTOR pathway30. In both the PC12 cell model of oxygen glucose deprivation and the rat brain model of permanent focal ischemia, IPC induced the activation of autophagy, demonstrating that the generation and degradation of autophagosomes was increased after I/R38. However, the inhibition of autophagic activity by 3-MA or Baf A1 attenuated the neuroprotective effect of IPC. Furthermore, rapamycin reduced the cerebral ischemic damage induced by the permanent focal ischemia.
Mitochondria and autophagy
Mitochondria are subcellular units that produce ATP via the oxidative phosphorylation pathway and play an important role in cell death and survival. Mitochondria undergo continuous remodeling through cycles of fusion and fission to maintain a high functional quality. The damaged and dysfunctional mitochondria that exhibit low membrane potential and generate excessive ROS are degraded through autophagy, namely mitophagy39. A large number of autophagosomes containing mitochondria have been observed during myocardial ischemia-reperfusion injury40. In addition, mitochondria play a role in up-regulating autophagy in cells. For example, a large amount of mitochondrial fragmentation, which occurred prior to the autophagic process, was found in HL-1 cells exposed to ischemia-reperfusion injury29. Recently, it has been demonstrated that mitophagy is up-regulated during ischemic injury41. Because mitochondria release pro-apoptotic factors and cytochrome c and produce a large amount of ROS after ischemia-reperfusion, which can lead to apoptosis and necrosis, the up-regulation of mitophagy may have a beneficial effect in ischemic injury42.
Several studies have reported that mitochondrial fragmentation and mitochondrial fission are required for inducing mitophagy43. Mitochondrial fission occurred before the up-regulation of autophagy during ischemic injury, whereas the inhibition of mitochondrial fission induced by ischemia reduced the increase in autophagy44. In addition, a study demonstrated that opening the mitochondrial permeability transition pore (mPTP) induced mitophagy after ischemia-reperfusion in cardiomyocytes, whereas inhibiting the mPTP using cyclosporine A, a mitochondrial inhibitor, attenuated the activation of autophagy, suggesting that mPTP acts as an upstream regulator for mitophagy in ischemic injury45. Mitochondrial integrity and the maintenance of energy are important for IPC-induced protection against I/R injury. Purportedly, the mechanism of IPC is associated with the mild depolarization of the mitochondrial inner membrane to open the mitochondrial ATP-sensitive potassium channel (mitoKATP) or transiently open the mPTP.It has been proposed that mitochondrial depolarization is a signal for the removal of damaged mitochondria via autophagy. Bnip3, a member of the BH3-only proteins, not only induces apoptosis but also serves as an essential regulator in mitophagy46. Bnip3 triggers a protective stress response with upregulation of autophagy and removal of damaged mitochondria during I/R injury. It has been reported that the up-regulation of mitophagy mediated by hypoxia is associated with the hypoxia-dependent factor-1-dependent expression of Bnip347. Recent studies have demonstrated that a number of proteins also play a key role in the activation of mitophagy, such as the PTEN-induced putative kinase protein-1 (Pink1) and parkin. It has been reported that parkin induced mitophagy in cells48; meanwhile, parkin was translocated to the mitochondria through Pink149.
The endoplasmic reticulum stress and unfolded protein response
The endoplasmic reticulum (ER) plays a critical role in correct protein synthesis and the folding and maintenance of Ca2+ homeostasis. The disturbance of the ER environment or function can induce ER stress, leading to the accumulation of unfolded or misfolded proteins. ER stress can activate the unfolded protein response (UPR) as a compensatory response, which protect the cells from damage through the activation of multiple functions, such as inducing the degradation of misfolded proteins and the expression of ER chaperone proteins50. Several studies have reported that ischemia-reperfusion activated the UPR in mouse hearts51. The misfolded proteins that accumulate during ER stress are degraded via proteasomes, and excessive unfolded proteins are degraded via autophagy. There is evidence that ER stress induces autophagy. In contrast, the inhibition of autophagy results in cell death mediated by ER stress52. In addition, the activation of autophagy elicited by ER stress is associated with UPR. For example, autophagy was up-regulated by tunicamycin, and thapsigargin acted as an ER stressor in neuroblastoma cells. ER chaperones, such as GRP78 and BiP, also regulate the autophagy induced by ER stress. The knockdown of GRP78 by siRNA inhibited the up-regulation of autophagy induced by ER stress in mammalian cells53. PERK/eIF2α phosphorylation, an important mediator of the UPR, is involved in the transition of LC3 I to LC3 II in cells54. Moreover, the release of ER calcium was required for the activation of autophagy55. Additionally, ER stress is directly linked to autophagy. It has been shown that Atg1 activity is enhanced when autophagy is induced by ER stress. Furthermore, Beclin1 is localized primarily at the ER52. ER stress is initiated during I/R injury, leading to the accumulation of unfolded or misfolded protein, thereby disrupting cellular homeostasis and resulting in apoptotic cell death. Studies performed on isolated rat heart and primary cultured cortical neurons demonstrated that the increased autophagy induced by IPC suppressed the excessive ER stress during I/R56. 3-MA attenuated neuroprotection by IPC and significantly inhibited the activation of autophagy and ER chaperones, such as GRP78, induced by IPC. Moreover, the ER stress inhibitor recovered the neuroprotection of IPC in the presence of 3-MA. The phosphorylation or activation of eIF2a and PERK are involved in ER stress in response to I/R injury.
Reactive oxygen species (ROS)
Oxidative stress and increased reactive oxygen species (ROS) are primary mediators of ischemia-reperfusion injury57, and in turn, ischemia-reperfusion contributes to the mitochondrial damage that generates excess ROS. Recently, it has been reported that lipopolysaccharide (LPS) treatment enhanced ROS and activated autophagy in neonatal cardiomyocytes58. Furthermore, evadiamine, a quinozole alkaloid, induced autophagy through the increase in ROS in HeLa cells59, which suggests that ROS production is involved in the regulation of autophagy. On the one hand, ROS induces autophagy via mitochondrial damage; a study demonstrated that the inhibition of mitochondrial electron transport chain (mETC) complexes I and II up-regulated autophagy and increased ROS in U87 and HeLa cells. Conversely, treatment with a ROS scavenger inhibited the increased autophagy by inhibiting the mETC complexes60. On the other hand, ROS also induces autophagy through direct oxidative modifications of the autophagy pathway. For example, H2O2 inhibited cysteine protease Atg4 activity, which promoted ATG8-PE conjugation and LC3 conversion, thereby inducing autophagy61. In addition, because Beclin1 expression is dependent on its interaction with Bcl-2, Beclin1 plays a role in the autophagy induced by ROS, which is associated with the expression of Bcl-2. ROS-induced autophagy is associated with cell survival and cell death, which is dependent upon ROS production. In our studies, we have demonstrated that hyperbaric oxygen preconditioning protects the brain from subsequent ischemia-reperfusion injury. The up-regulation of autophagy is involved in the neuroprotection of hyperbaric oxygen preconditioning in a rat model of focal cerebral ischemia8. Furthermore, this neuroprotective effect is mediated by an appropriate increase in endogenous ROS in vivo and in vitro62,63. These data suggest that the neuroprotective mechanism of autophagy activation by HBO preconditioning is mediated by the generation of ROS.
Apoptosis and autophagy
In general, it is accepted that autophagy and apoptosis are completely different processes. However, increasing evidence has demonstrated that autophagy and apoptosis are induced by common regulators and signaling pathways. Moreover, cooperation exists between autophagy and apoptosis64. For example, Atg5 not only has an important effect on autophagosome formation but also inhibits apoptosis. It has been reported that the knockdown of Atg5 inhibits autophagy, which induces apoptosis65. Moreover, the inactivation of Atg5 mediated by calpain enhanced cytochrome c release and caspase activity66, suggesting that the effect of Atg5 inactivation on apoptosis is similar to the effect of the pro-apoptotic proteins belonging to the Bcl-2 family. Additionally, Atg5 interacts with Bcl-XL, leading to apoptosis, which indicates that Atg5 may promote apoptosis through a secondary apoptotic pathway. In addition, the Bcl-2 binding to Beclin1 also regulates autophagy and apoptosis. It has been reported that the Bcl-2 interaction with Beclin1 inhibits autophagy, and autophagy was inhibited through the overexpression of Bcl-2 in murine heart cells, indicating that Bcl-2 serves as a negative regulator of autophagy through the inhibition of Beclin164. The BH3 domain of Beclin1 is involved in the binding of Bcl-2 and Beclin1, and a published study demonstrated that the overexpression of a Beclin1 mutant that cannot interact with Bcl-2 induces increased autophagy in the mouse heart67. Therefore, Bcl-2 not only plays an important role in apoptosis but also maintains autophagy levels to promote cell survival rather than cell death. Studies have demonstrated that Bcl-2 negatively regulates the activation of the Beclin1-class III PI3K complex; when the association of Bcl-2 with the Beclin1-class III PI3K complex was interrupted, autophagy was up-regulated in response to stress33. Caspase mediates the cleavage of Beclin1, which induces autophagy; furthermore, a recent study demonstrated that the cleavage of Beclin1 by caspase promoted the production of pro-apoptotic molecules from mitochondria, leading to apoptosis68.
In a cerebral hypoxia-ischemia model of neonatal rat, Carloni et al determined that the up-regulation of autophagy plays a neuroprotective role in cerebral ischemia. Beclin1 was markedly enhanced after hypoxia-ischemia and was co-localized with the TUNEL-positive cells, suggesting that the up-regulation of autophagy and promotion of apoptosis occurred simultaneously during ischemic injury. Moreover, inhibiting the activation of autophagy caused rapid cell necrosis by interrupting the progress of apoptosis30. The relationship between autophagy and apoptosis was identified in another study using a rat model of severe perinatal asphyxia69. The expression of Bcl-2 was detected in the liver following IPC, which co-localized with Beclin1. Increased Bcl-2 expression was associated with decreased Beclin 1 and up-regulated LC3-II. In addition, necrosis was reduced by IPC in the liver; however, the TUNEL assay and the expressions of caspase 3, 8, and 9 did not change. These data suggest that the activation of autophagy is involved in the IPC-induced protection against I/R through the interaction of Bcl-2 with Beclin1 and the limitation of necrosis in the human liver70.
The studies also demonstrated that 3-MA significantly suppressed the IPC-induced increase in heat shock protein 70 (HSP70) after ischemia, suggesting that the up-regulation of HSP70 participates in the autophagic mechanism underlying the IPC-induced neuroprotection. HSP70 serves as a chaperone and participates in two protein degradation systems, the autophagy/lysosomal pathway and the ubiquitin-proteasomal pathway71. Similarly, the involvement of autophagic activity in ischemic tolerance in IPC has been demonstrated in the heart72,73. Meanwhile, it has been shown that autophagy induced by IPC is essential for cardioprotection74. BAG-1 is a multifunctional molecule involved in cell survival that protects cardiomyocytes through the inhibition of apoptosis in association with Hsp70 and the Hsc70 chaperone during I/R injury75. A study demonstrated that the cardioprotection elicited by IPC is mediated by the activation of autophagy in association with BAG-140,76. Chemotherapy can aggravate I/R injury during liver surgery; IPC reduced the damage in livers previously treated with chemotherapy. Despite of the classic IPC, there is increasing evidence that agent preconditioning, such as rapamycin and, 2-chloro-N(6)-cyclopentyladenosine (CCPA, an adenosine A1 receptor agonist) mimics the protective effect of IPC in I/R through the significant up-regulation of autophagy77,78. We demonstrated the essential role for autophagy in a study using a rat model of transient focal cerebral ischemia by middle cerebral artery occlusion; we determined that the activation of autophagy is associated with the neuroprotection of hyperbaric oxygen preconditioning against cerebral I/R injury8.
It is conceivable that autophagy is required for the protection elicited by IPC. However, there is an alternative view. For example, Wu et al demonstrated that prolonged ischemia induced renal dysfunction in rat through the promotion of apoptosis and autophagy. Following a brief intermittent reperfusion during prolonged ischemia, IPC inhibited the apoptosis and autophagy induced by I/R and reduced renal dysfunction, suggesting that the activation of autophagy plays a negative role in IPC-induced protection79. Further investigations will be required to address this possibility.
Conclusion
Increasing evidence suggests that the activation of autophagy is involved in IPC protection against I/R injury in the body. However, autophagy is a double-edged sword. Excessive autophagic activity can damage normal proteins and organelles resulting in programmed cell death. Therefore, many questions remain regarding the specific mechanism of how and under what conditions the activation of autophagy regulates the protective effect of IPC. Not all factors that induce autophagy are beneficial. For example, intracellular Ca2+ overload because of ischemic injury also induces autophagy, which represents damage rather than protection. Moreover, further studies will be needed to determine the optimum autophagic activity needed to obtain maximal benefits during I/R injury. In addition, it has been demonstrated that the activation of autophagy plays an important role in the ischemic tolerance of pharmaceutical or non-ischemic preconditioning to I/R, suggesting that autophagy may be a potential target for the treatment or prevention of organ and tissue injuries by I/R. More studies will be required to elucidate the autophagic cell survival pathways and may allow the clinic application of autophagy for therapeutic purposes.

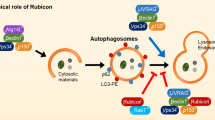
Schematic model of autophagy pathways involved in the protective effect of ischemic preconditioning. Ischemic preconditioning causes moderately energy depletion, production of ROS and ER stress, which in turn up-regulate autophagic activity and promote cellular survival. Signaling pathways include activation of AMPK, Beclin1 and class-III PI3K, and inhibition of mTOR and class I PI3K.
References
Levine B, Klionsky DJ . Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6: 463–77.
Mizushima N . Autophagy: process and function. Genes Dev 2007; 21: 2861–73.
Huang J, Klionsky DJ . Autophagy and human disease. Cell Cycle 2007; 6: 1837–49.
Murry CE, Jennings RB, Reimer KA . Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124–36.
Bonventre JV . Kidney ischemic preconditioning. Curr Opin Nephrol Hypertens 2002; 11: 43–8.
Kirino T . Ischemic tolerance. J Cereb Blood Flow Metab 2002; 22: 1283–96.
Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH . Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6: 482–94.
Yan W, Zhang H, Bai X, Lu Y, Dong H, Xiong L . Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res 2011; 1402: 109–21.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93.
Yang Z, Klionsky DJ . Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12: 814–822.
Kiffin R, Christian C, Knecht E, Cuervo AM . Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15: 4829–40.
Mijaljica D, Prescott M, Devenish RJ . Mitophagy and mitoptosis in disease processes. Methods Mol Biol 2010; 648: 93–106.
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182: 685–701.
Geng J, Nair U, Yasumura-Yorimitsu K, Klionsky DJ . Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell 2010; 21: 2257–69.
Reggiori F, Shintani T, Nair U, Klionsky DJ . Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 2005; 1: 101–9.
Hoyer-Hansen M, Jaattela M . Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14: 1576–82.
Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12–Apg5 conjugate. J Cell Sci 2003; 116: 1679–88.
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152: 657–68.
Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet 2005; 37: 771–6.
Webb JL, Ravikumar B, Rubinsztein DC . Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 2004; 36: 2541–50.
Eskelinen EL . Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005; 1: 1–10.
Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol 2005; 167: 1713–28.
Mousavi SA, Brech A, Berg T, Kjeken R . Phosphoinositide 3-kinase regulates maturation of lysosomes in rat hepatocytes. Biochem J 2003; 372: 861–9.
Takagi H, Matsui Y, Hirotani S, Sakoda H, Asano T, Sadoshima J . AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy 2007; 3: 405–7.
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100: 914–22.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–41.
Wang C, Wang Y, McNutt MA, Zhu WG . Autophagy process is associated with anti-neoplastic function. Acta Biochim Biophys Sin 2011; 43: 425–32.
Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by Beclin1, a novel Bcl-2 interacting protein. J Virol 1998; 72: 8586–96.
Hamacher-Brady A, Brady NR, Gottlieb RA . Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281: 29776–87.
Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W . Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010; 6: 366–77.
Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, et al. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol 2006; 40: 846–52.
Obara K, Sekito T, Ohsumi Y . Assortment of phosphatidylinositol 3-kinase complexes — Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell 2006; 17: 1527–39.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8: 688–99.
Gu Y, Wang C, Cohen A . Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Lett 2004; 577: 357–60.
Zeng X, Overmeyer JH, Maltese WA . Functional specificity of the mammalian Beclin-Vps34 PI3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 2006; 119: 259–70.
Aki T, Yamaguchi K, Fujimiya T, Mizukami Y . Phosphoinositide 3-kinase accelerates autophagic cell death during glucose deprivation in the rat cardiomyocyte-derived cell line H9c2. Oncogene 2003; 22: 8529–35.
Takemura G, Maruyama R, Goto K, Kanamori H, Tsujimoto A, Minatoguchi S, et al. Fate of isolated adult cardiomyocytes undergoing starvation-induced autophagic degeneration. Autophagy 2009; 5: 90–2.
Park HK, Chu K, Jung KH, Lee ST, Bahn JJ, Kim M, et al. Autophagy is involved in the ischemic preconditioning. Neurosci Lett 2009; 451: 16–9.
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27: 433–46.
Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK . Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med 2009; 13: 373–87.
Elmore SP, Qian T, Grissom SF, Lemasters JJ . The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 2001; 15: 2286–7.
Degli ED, Sebagh M, Pham P, Reffas M, Pous C, Brenner C, et al. Ischemic preconditioning induces autophagy and limits necrosis in human recipients of fatty liver grafts, decreasing the incidence of rejection episodes. Cell Death Dis 2011; 2: e111.
Brunk UT, Terman A . The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 2002; 269: 1996–2002.
Brady NR, Hamacher-Brady A, Gottlieb RA . Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim Biophys Acta 2006; 1757: 667–78.
Halestrap AP, Clarke SJ, Javadov SA . Mitochondrial permeability transition pore opening during myocardial reperfusion — a target for cardioprotection. Cardiovasc Res 2004; 61: 372–85.
Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF . BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27: 6229–42.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–903.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8: e1000298.
Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010; 6: 871–8.
Rutkowski DT, Kaufman RJ . A trip to the ER: coping with stress. Trends Cell Biol 2004; 14: 20–8.
Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC . Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res 2006; 99: 275–82.
Yorimitsu T, Klionsky DJ . Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 2007; 3: 160–12.
Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS . The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 2008; 15: 1460–71.
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14: 230–9.
Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA . The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J 2007; 274: 3184–97.
Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 2012; 8: 310–25.
Vanden HT, Li C, Shao Z, Schumacker PT, Becker LB . Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol 1997; 29: 2571–83.
Khadour FH, Panas D, Ferdinandy P, Schulze C, Csont T, Lalu MM, et al. Enhanced NO and superoxide generation in dysfunctional hearts from endotoxemic rats. Am J Physiol Heart Circ Physiol 2002; 283: H1108–15.
Yang J, Wu LJ, Tashino S, Onodera S, Ikejima T . Reactive oxygen species and nitric oxide regulate mitochondria-dependent apoptosis and autophagy in evodiamine-treated human cervix carcinoma HeLa cells. Free Radic Res 2008; 42: 492–504.
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB . Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 2007; 120: 4155–66.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z . Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26: 1749–60.
Li Q, Li J, Zhang L, Wang B, Xiong L . Preconditioning with hyperbaric oxygen induces tolerance against oxidative injury via increased expression of heme oxygenase-1 in primary cultured spinal cord neurons. Life Sci 2007; 80: 1087–93.
Nie H, Xiong L, Lao N, Chen S, Xu N, Zhu Z . Hyperbaric oxygen preconditioning induces tolerance against spinal cord ischemia by upregulation of antioxidant enzymes in rabbits. J Cereb Blood Flow Metab 2006; 26: 666–74.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–52.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–40.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124–32.
Pattingre S, Levine B . Bcl–2 inhibition of autophagy: a new route to cancer? Cancer Res 2006; 66: 2885–8.
Wirawan E, Vande WL, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010; 1: e18.
Ginet V, Puyal J, Clarke PG, Truttmann AC . Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol 2009; 175: 1962–74.
Domart MC, Esposti DD, Sebagh M, Olaya N, Harper F, Pierron G, et al. Concurrent induction of necrosis, apoptosis, and autophagy in ischemic preconditioned human livers formerly treated by chemotherapy. J Hepatol 2009; 51: 881–9.
Meriin AB, Sherman MY . Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia 2005; 21: 403–19.
Dosenko VE, Nagibin VS, Tumanovska LV, Moibenko AA . Protective effect of autophagy in anoxia-reoxygenation of isolated cardiomyocyte? Autophagy 2006; 2: 305–6.
Dosenko VE, Nagibin VS, Tumanovskaya LV, Zagoriy VY, Moibenko AA, Vaage J . Proteasome inhibitors eliminate protective effect of postconditioning in cultured neonatal cardiomyocytes. Fiziol Zh 2006; 52: 15–24.
Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RJ, et al. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 2010; 3: 365–73.
Townsend PA, Cutress RI, Carroll CJ, Lawrence KM, Scarabelli TM, Packham G, et al. BAG-1 proteins protect cardiac myocytes from simulated ischemia/reperfusion-induced apoptosis via an alternate mechanism of cell survival independent of the proteasome. J Biol Chem 2004; 279: 20723–8.
Gurusamy N, Lekli I, Gherghiceanu M, Popescu LM, Das DK . BAG-1 induces autophagy for cardiac cell survival. Autophagy 2009; 5: 120–1.
Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC . Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 2006; 41: 256–64.
Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RJ, et al. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol 2009; 104: 157–67.
Wu HH, Hsiao TY, Chien CT, Lai MK . Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J Biomed Sci 2009; 16: 19.
Acknowledgements
This work was supported by the National Science Foundation for Distinguished Young Scholars (Grant 30725039) and the key project for the National Natural Science Foundation of China (Grant 30930091).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Yan, Wj., Dong, Hl. & Xiong, Lz. The protective roles of autophagy in ischemic preconditioning. Acta Pharmacol Sin 34, 636–643 (2013). https://doi.org/10.1038/aps.2013.18
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.18
Keywords
This article is cited by
-
Activation of Autophagy in Human Uterine Myometrium During Labor
Reproductive Sciences (2020)
-
Zearalenone altered the cytoskeletal structure via ER stress- autophagy- oxidative stress pathway in mouse TM4 Sertoli cells
Scientific Reports (2018)
-
Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination
Cell Death & Differentiation (2017)
-
The critical roles of mitophagy in cerebral ischemia
Protein & Cell (2016)
-
Autophagy Promotes Peripheral Nerve Regeneration and Motor Recovery Following Sciatic Nerve Crush Injury in Rats
Journal of Molecular Neuroscience (2016)
