
Abstract
The ROCO proteins are a family of large, multidomain proteins characterised by the presence of a Ras of complex proteins (ROC) domain followed by a COR, or C-terminal of ROC, domain. It has previously been shown that the ROC domain of the human ROCO protein Leucine Rich Repeat Kinase 2 (LRRK2) controls its kinase activity. Here, the ability of the ROC domain of another human ROCO protein, Death Associated Protein Kinase 1 (DAPK1), to bind GTP and control its kinase activity has been evaluated. In contrast to LRRK2, loss of GTP binding by DAPK1 does not result in loss of kinase activity, instead acting to modulate this activity. These data highlight the ROC domain of DAPK1 as a target for modifiers of this proteins function and casts light on the role of ROC domains as intramolecular regulators in complex proteins with implications for a broad range of human diseases.
Similar content being viewed by others


Introduction
The human ROCO proteins are defined by the presence of a GTPase domain, termed ROC for RAS of complex proteins, followed by a COR (c-terminal of ROC) domain1. The family consists of four members: Leucine Rich Repeat Kinases 1 and 2 (LRRK1 and LRRK2), Death Associated Protein Kinase 1 (DAPK1) and MFH-amplified sequences with Leucine Rich tandem repeats 1 or MASL12. All four have links to human disease: mutations in LRRK2 are the most common genetic cause of Parkinson’s disease (PD)3 and all of the human ROCO proteins have been linked to cancer2. More recently, genome wide association studies have identified the LRRK2 locus as being involved in susceptibility to sporadic PD, Crohn’s disease and leprosy4,5,6. With regard to DAPK1 in particular, alterations in the expression and activity of this protein are associated with changes in cell proliferation and control of cell death7,8 and both silencing and mutations of DAPK1 altering its expression have been linked to chronic lymphocytic Leukaemia9. Because of this, DAPK1 has been highlighted as a potential therapeutic target in cancer10,11. In addition to its links with cancer, several studies have suggested that DAPK1 may play a role in Alzheimer’s disease, although these data remains controversial12,13. A genetic link between DAPK1 and PD has been examined, with no evidence that genetic variability in the DAPK1 gene alters the risk of developing the disease14.
To understand how this family of complex, multidomain proteins operates in a cellular context and how their dysfunction leads to disease, it is important to gain an insight into how their individual domains relate to one another. Structural studies would be the most informative approach, describing the precise spatial arrangement of domains, however to date only fragments of the ROCO proteins have been purified to the point where crystallographic studies have been feasible and have provided only partial information as to domain/domain interactions15,16,17,18. Three of the ROCO proteins contain kinase effector domains (LRRK1, LRRK2 and DAPK1). In all three cases, targeted mutagenesis of individual domain activities has proved revealing as to how the various domains relate to each other – in the case of LRRK1 and LRRK2, disruption of the GTP binding properties of the ROC domain have been reported to have a dramatic impact on the kinase activities of these proteins19,20. This has led to the suggestion that the ROC and kinase domains in these proteins act in a manner analogous to the small GTPases and kinase such as Ras and Raf, with the ROC domain acting to regulate the kinase activity21. Ablation of the kinase activity of DAPK1 results in a decrease in the action of the Death domain of this protein22. Similar to this, artificial lowering of LRRK2’s kinase activity either by mutations or inhibitors has been shown to decrease toxicity associated with PD linked mutations23,24,25. Because of the data linking the kinase activities of LRRK2 and DAPK1 to cell death, the kinase domains of these proteins have been identified as potential therapeutic targets in cancer and PD respectively26,27. Bioinformatic analysis of the amino acid sequences of the kinase domains of the human LRRK genes and DAPK1 has revealed that they belong to different kinase families28. Furthermore, the kinase domain of DAPK1 is N-terminal of the ROC-COR module whereas in LRRK1 and 2 the kinase domains are at the C-terminus suggesting distinct structural interactions. Coupled to data revealing that the ROC and COR domains of these three proteins are closely related, this has led to the suggestion that the kinase domains of the LRRK proteins transferred to the ROCO family in a separate evolutionary event to the transfer of kinase domain of DAPK129. This, in turn, raises the possibility that the intramolecular regulation of kinase activity is different in DAPK1 compared to the LRRK proteins. A recent study by Carlessi et alia examined these issues, demonstrating that the ROC domain of DAPK1 is indeed a GTP binding domain and that loss of GTP binding acts to increase, rather than decrease, the kinase activity of DAPK1 due to loss of an inhibitory phosphorylation event30. The data presented in this study replicate that reported by Carlessi et alia, further supporting the thesis that the ROC domain of DAPK1 can bind GTP and examines how the loss of GTP binding impacts on the kinase activity and the ability of this protein to form a complex under native conditions.
Results
To examine the role of the ROC domain in the function of DAPK1, an artificial mutation - T701N - was generated in the ROC domain of DAPK1, designed to exclude GTP from its binding pocket based upon sequence homology to equivalent residues in LRRK2 which have been demonstrated to prevent GTP binding19,31,32,33,34 (Figure 1A-C). This mutant was used to investigate the GTP binding properties of DAPK1 in comparison with the wild type protein. HA tagged wild type DAPK1 is capable of binding to GTP immobilized on sepharose beads, an interaction that can be interrupted by the addition of a molar excess of GTP (figure 2A). As predicted, the T701N mutation is unable to bind the GTP resin thus acting to disrupt the binding of DAPK1 to GTP (figure 2A). To examine whether endogenous DAPK1 displays the same properties as epitope tagged DAPK1, HEK293T cell lysates were examined in the same way and, using an antibody specific for DAPK1, both binding to GTP sepharose and disruption of this interaction upon addition of a molar excess of GTP were apparent (figure 2B), with enrichment of DAPK1 in the precipitated fractions. This data verifies the GTP binding properties of DAPK1 at the endogenous level.

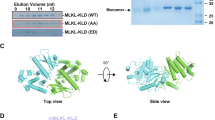
(A) Sequence alignment of DAPK1 and LRRK2 (left panel) and DAPK1 and the prokaryotic ROCO protein from Chlorobium tepidum (right panel) showing shared residues.(B) Crystal structure of the Chlorobium tepidum ROCO protein ROC-COR fragment (co-ordinates taken from reference 16, PDB reference 3PDU) showing dimeric complex (monomer one coloured blue, monomer two coloured turquoise). The GxxxxGKT motif in the GTP binding pocket is highlighted in red for monomer one. (C) Magnified view of the GTP binding pocket of the Chlorobium tepidum ROCO protein showing the T644 residue, equivalent to the T701 residue in DAPK1.

(A) GTP binding by wild type and GTP binding dead mutated HA-tagged DAPK1, showing input lysates (lower panel) and DAPK1 precipitated by binding to GTP-agarose beads.The T701N mutated form of DAPK1 was unable to bind GTP-agarose. The interaction of wild type DAPK1 with GTP could be disrupted by the addition of a molar excess of GTP. (B) Interaction of endogenous DAPK1 expressed in HEK293 cells with GTP.
Having established that DAPK1 is a GTP binding protein, the impact of GTP binding upon the kinase domain was investigated. To assess this, the kinase activity of immunoprecipitated DAPK1 was measured in an in vitro kinase assay as previously described25 (figure 3A). A K42W mutation, which is predicted to interfere with the ability of DAPK1 to bind ATP35, was used as control to exclude the possibility that any 32P incorporation observed was not due to heterologous kinases co-precipitating with DAPK1. There was no observable impact of GTP binding properties of DAPK1 (as modulated by the T701N artificial mutation) upon its kinase activity, compared to wild type protein as measured by autophosphorylation (quantified data normalized to immunoprecipitated protein shown in figure 3B). In contrast, the T701N mutation resulted in a significant increase in activity compared to wild type protein as measured by the phosphorylation of the generic kinase substrate Myelin Basic Protein (figure 3C). Relative autophosphorylation was also measured in the absence of a generic substrate, again with no significant difference between wild type and T701N kinase activity (data not shown).

(A) Representative autoradiograph and immunoblot analysis of the kinase activity of wild type and T701N forms of DAPK1.The upper panel shows autophosphorylation activity as represented by incorporation of 32P. The middle panel shows immunoreactivity for HA tagged DAPK1 immunoprecipitated for the assay. The lower panel shows phosphorylation of Myelin basic protein by DAPK1. B) Quantified phosphorylation is displayed corrected for total protein as quantified by immunoblot (mean and standard error displayed). Autophosphorylation levels were equivalent in the wild type and T701N assays and both were significantly different (n = 3, 1 way ANOVA with Tukeys post hoc correction) to the K42W assay. (C) Phosphorylation of MBP (quantified phosphorylation, arbitrary units). T701N displayed significantly higher phosphorylation than wild type protein (n = 3, 1 way ANOVA with Tukeys post hoc correction).
To investigate whether the T701N mutation has an impact on the cellular localisation of DAPK1, HEK293T cells transfected with wild type and T701N DAPK1 were examined by immunocytochemical labelling (figure 4). Wild type, K42W and T701N DAPK1 displayed similar patterns of staining.

Immunocytochemical analysis of DAPK1 wild type (A) K42W (B) and T701N (C) expression in HEK293T cells.
No significant alteration in cellular localisation was observed due to the insertion of the artificial mutations. Scale bar represents 20 μm in main image, 10 μm in magnified view.
DAPK1 and LRRK2 have been demonstrated to form complexes consistent with functioning as a dimer and so to examine the impact of alterations in enzymatic activity upon the ability of DAPK1 to form a dimer, size exclusion chromatography (SEC) was undertaken with cell lysates from cells transfected with the HA tagged wild type, kinase dead and GTP binding dead forms of the protein respectively (figure 5A quantified average data, 5B representative dot blot and 5C representative western blot). In contrast to LRRK2, loss of GTP binding or kinase activity of DAPK1 does not result in disruption of an individual chromatographic peak. However, while the wild type and the K42W forms eluted at ≈ 400 kDa consistent with a dimeric complex, the T701N mutation caused the protein to fractionate along a broader range of molecular weights. To use an alternative approach to examine complex formation by DAPK1, blue native gel electrophoresis was undertaken with cell lysates expressing the wild type, T701N and K42W mutants (Figure 5D). Bands consist with the observed peaks from SEC analysis were present at ≈ 400 kDa for wild type, K42W and T701N. However, the T701N mutant also exhibits an unresolved smear above the 400 kDa band, consistent with the SEC results.

(A) Size exclusion chromatography analysis of DAPK1 wild type, K42W and T701N. Data represents average of 3 independent experiments, with independent replicates shown by individual dots.(B) Representative dot blots for wild type, K42W and T701N DAPK1. (C) Representative western blots of peak fractions for wild type, K42W and T701N DAPK1. (D) Blue native gel electrophoresis analysis of wild type, K42W and T701N DAPK1.
Finally, immunogold electron microscopy (EM) was used to examine the oligomeric state of DAPK1 (figure 6). Lysates from HEK293T cells transiently transfected with HA-DAPK1 wild type were subjected to SEC and the fraction corresponding to the 400 kDa peak diluted 20 times and analysed by immunogold EM. As shown in figure 6A, gold-labelled particles display a pattern of doublets that are consistent with a dimeric DAPK1 complex. Gold particles were occasionally observed organized in triplets or even higher order complexes, suggesting that oligomers larger than dimers may co-exist. To rule out that these apparent DAPK1 dimers are not due to a stochastic event, the frequency distribution of particle distances was calculated and normalized by the annulus area within which the particles were counted, since the probability of finding a particle within a given annulus increases in proportion to the area of that annulus. The distribution frequency plot of particle distances measured from different EM fields (n = 559) indicates a clear peak around 20 nm supporting the thesis that DAPK1 is able to form dimers and, possibly, higher order oligomers (figure 6B). The T701N mutant was also investigated using immunogold labelling. Similar to the wild-type, gold labelled doublets were observed in the 400 kDa fraction of DAPK1-T701N (quantified data shown in figure 6C) but a significantly smaller number of total gold particles were found compared to the wild-type protein (an average of 29 gold particles per micrograph for the wild-type compared to 11 gold particles per micrograph for the T701N mutant). This is consistent with the observation that this mutant also fractionates at higher molecular weights and is therefore spread among a larger number of fractions compared to the wild-type under conditions of equal total protein (as shown in figure 5C).

Transmission electron microscopy analysis of DAPK1 overexpressing cell lysates.
(A) Representative EM image of immunogold labeled samples from chromatographic fractions corresponding to the 400 kDa peak. Bar is 50 nm. (B) Analysis of frequency distributions of distances among gold particles for wild type DAPK1 normalized by the annulus area, with the annulus thickness corresponding to the bin size (5 nm) (C) Analysis of frequency distribution for T701N DAPK1 normalized by the annulus area, with the annulus thickness corresponding to the bin size (5 nm).
Discussion
The data from this study demonstrates that DAPK1 is a GTP binding protein, that the GTP binding properties of its ROC domain act to modulate the kinase activity of this protein and that DAPK1 forms a complex consistent with a dimeric active conformation. This data replicates and extends the recent study by Carlessi and co-workers examining the role of the ROC domain of DAPK1 in regulating protein function30.
To investigate whether DAPK1 has a functional ROC domain two approaches were used: competitive binding using GTP sepharose and GTP free in solution and a genetic approach using a targeted point mutation in the predicted GTP binding pocket of the DAPK1 ROC domain. GTP binding was demonstrated using HA tagged and endogenous DAPK1, with the T701N mutation prevented binding of HA tagged DAPK1 to GTP sepharose beads. It is possible that the introduction of the T701N mutation into the GTP binding pocket of DAPK1 and the subsequent exclusion of GTP, results in a destabilisation of the tertiary and quaternary structure of the protein. Although it was not possible to directly examine whether DAPK1 containing the T701N mutation is correctly folded, several indirect strands of evidence suggest that the T701N mutation does not result in the misfolding and aggregation of DAPK1. First, steady state expression of DAPK1 is not altered by the introduction of the T701N mutation. If the T701N mutation destabilised DAPK1, it would be expected that steady state expression would be lower as the protein is targeted for disposal. Secondly, both wild type and T701N DAPK1 display similar cytoplasmic expression patterns – if the T701N mutation destabilised DAPK1 as a whole a possible outcome of this would be its localisation to inclusion bodies as the cell attempts to remove the misfolded protein. Finally, the T701N form of DAPK1 has a catalytically active kinase domain, demonstrating that the kinase domain is correctly folded. Whilst this does not provide evidence regarding the folded state of the other domains in DAPK1, it suggests that the T701N mutation does not result in a complete destabilisation of the holoprotein.
With regard to the impact of the T701N mutation on kinase activity, it is notable that, in contrast to LRRK2, the loss of GTP binding for DAPK1 does not result in an overall decrease in autophosphorylation. Intriguingly, the T701N mutation increases the activity of DAPK1 towards a generic substrate, MBP. This is consistent with the increase in DAPK1 kinase activity towards Myosin Light Chain described by Carlessi and co-workers. This suggests that the action of the ROC domain as an intramolecular regulator is more complex than a simple on/off switch as initial data from the leucine rich repeat kinases suggested, whilst highlighting the importance of this domain in the regulation of kinase activity21 and its potential as a drug target. Recent data investigating the biology of the ROC domain in regulating LRRK2 has highlighted the complex nature of the regulatory role of this domain34,36, with a study investigating the role of the WD40 domain of LRRK2 on the kinase activity of the protein further emphasising the multiple levels of regulation that govern ROCO protein biology37.
There are a number of strands of evidence that suggest that the ROCO proteins are functionally active as dimeric complexes15,38,39,40,41,42, although this has been re-examined in a recent paper43. A previous study examined the ability of the human ROCO proteins to homo- and heterodimerise and reported that DAPK1 was able to form a homodimer and had the capacity to form heterodimers with LRRK1 and LRRK244. It has also been reported that loss of GTP binding and ablation of kinase activity, in LRRK2 can act to disrupt dimer formation38. The data in this study examining the impact of loss of GTP binding or kinase activity on the ability of DAPK1 to form a dimer contrasts markedly with data for LRRK2. Whereas loss of GTP binding/kinase activity results in the disruption of putative dimer formation in LRRK2, this is not the case for DAPK1. Loss of kinase activity has no significant impact on the apparent size of the complex formed by DAPK1, while the T701N mutation results in a protein that fractionates along a broader range of molecular weights. There are several possible reasons for this change in complex size as measured by SEC. First, it is possible that DAPK1 is in equilibrium among different oligomeric states (dimers, trimers, tetramers and/or higher order oligomers) with GTP binding favouring the dimeric conformation. This scenario is supported by the immunogold EM results, which highlight the presence of higher order oligomers other than the dimer (figure 6A). Loss of GTP binding alters the oligomeric state of DAPK1 shifting the equilibrium toward other conformers (trimers, tetramers or higher order oligomers). Second, the GTP binding could act to regulate the recruitment of a co-factor protein(s). As a consequence, loss of GTP binding would lock the protein into a bound state and therefore cause the apparent complex mass to increase. Finally, GTP could function as an allosteric regulator, with loss of GTP binding influencing the three dimensional conformation of the enzyme. Allosteric transitions are common among enzymes and are often associated with a shift between two structurally different states (for example, from a compact to a more elongated conformation)45,46. Therefore Apo-DAPK1 may possess a more open conformation, thereby increasing the hydrodynamic radius and the apparent relative molecular mass of the complex observed in the chromatogram. Although this hypothesis would be consistent with a model for ROCO function proposed by Gasper and co-workers40 it seems the less likely given that the T701N protein displays increased polydispersity rather than a clear shift in molecular weight. Determining which of these possibilities is correct will require further investigations.
To complement the SEC data, immunogold EM was performed to examine directly the quaternary structure of DAPK1 from the SEC peak. These data provide the first direct evidence that DAPK1 can form dimers and suggest that dimerization may be a common feature among ROCO proteins42. One possibility is that DAPK1 and ROCO proteins in general, function as G proteins activated by nucleotide dependent dimerization to modulate kinase activity40. If dimerization is a required step for GTP binding/hydrolysis but not for kinase activity, then the observation that the GTP dead mutant T701N, but not the K42W kinase dead mutant, displays a different SEC profile to the wild type protein supports this notion. The TEM data in this study, however, do not suggest a major disruption of quaternary structure, highlighting that this aspect of DAPK1 biology requires further investigation and that atomic resolution structural data covering multiple domains of DAPK1 is an important unmet need.
The data from this study replicates data previously reported by Carlessi and co-workers. Despite different experimental conditions (for example: different epitope tags – the current study used HA tagged DAPK1 whereas the majority of experiments in the Carlessi study used a flag tag – and different generic kinase substrates – myelin basic protein in this study, maltose binding protein in Carlessi et alia), the results described above are broadly consistent with the previous study. In particular, the analysis of kinase activity and of complex formation for wild type and T701N DAPK1 reveal similar outcomes, suggesting that these are robust effects. Using blue native PAGE and TEM, this current study has extended the findings of Carlessi et alia regarding complex formation, supporting the conclusions reached using SEC.
In summary, this study confirms that DAPK1 is indeed a GTP binding protein and that loss of GTP binding results in an increase in phosphorylation activity directed towards a generic substrate compared to wild type protein, with no apparent increase in autophosphorylation demonstrating that there is an intramolecular regulation of function in DAPK1. SEC and TEM analysis of DAPK1 confirms that it exists under native conditions in a complex consistent with a dimeric structure and that loss of kinase activity does not alter this complex. Loss of GTP binding results in an increase of DAPK1 polydispersity, indicating that GTP binding may stabilize one conformation, i.e. the dimer. These data present a stark contrast with the impact of GTP binding in LRRK1 and LRRK2, two further members of the human ROCO protein family and highlight the complex regulation of the function of this family.
Methods
Constructs
N-terminal HA tagged DAPK1 constructs were obtained from Genecopoeia (www.genecopoeia.com). Site directed mutagenesis was carried out using the Quikchange II mutagenesis kit (Agilent) as described in the manufacturers instructions. Primer sequences are available from the authors upon request. All constructs were verified by sequencing prior to use.
Cell culture
HEK293T and COS7 cells were obtained from ATCC® (line identifiers CRL-1573 and CRL-1651 respectively) and cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) at 37°C in 5% CO2 prior to use.
Sequence alignment and structural modelling
Protein sequences were downloaded from RefSeq (http://www.ncbi.nlm.nih.gov/RefSeq/). Sequence alignments were carried out using protein BLAST (http://blast.ncbi.nlm.nih.gov/). Co-ordinates for the crystal structure of the Chlorobium tepidum ROCO protein (taken from reference 16, accession number 3PDU) were downloaded from the Protein Database (http://www.rcsb.org/pdb/home/home.do). Images were generated using Chimera (http://www.cgl.ucsf.edu/chimera/).
DAPK1 expression and GTP pulldown assays
HA-DAPK1 was transfected into COS7 cells using fugene 6 HD (Roche) as described in the manufacturers instructions. 24 hours after transfection, cells were harvested in lysis buffer G (100 mM Tris–HCl pH 7.5, 50 mM KCl, 5 mM MgCl2 1 mM EDTA, 0.1 mM DTT, 1% Triton X-100 and 1x complete protease inhibitor cocktail, Roche), lysed for 30 minutes at 4°C with rotation and precleared with 35 µl of 50% sepharose beads. Lysates were clarified by centrifugation at 10000 g at 4°C for 10 minutes and the supernatants used in subsequent experiments. DAPK1 expression was verified by immunoblotting following BCA assay (Pierce, as per manufacturers instructions), with 10 µg of lysate loaded onto 4-12% Bis Tris gels (Invitrogen) following denaturation in 4x sample buffer (Invitrogen) supplemented with 5% β-Mercaptoethanol (Sigma). Proteins were transferred to PVDF membrane (Millipore) and membranes blocked with 5% milk solution in Phosphate buffered saline for 1 hour prior to probing with primary (90 minutes) and secondary (60 minutes) antibodies. Membranes were washed three times in PBS supplemented with 1% tween and incubated with Pierce Supersignal ECL substrate. Membranes were exposed to Kodak biomax film and developed on a Kodak developer as per manufacturers instructions.
Kinase assays
kinase assays were carried out as previously described31. Briefly, DAPK1 constructs were transfected as above. Cells were isolated in cell signalling lysis buffer (Cell Signaling) plus protease inhibitors (Roche) and subjected to lysis in the presence of 35 µl of sepharose beads for 30 minutes with rotation at 4°C. Following clarification of lysates by centrifugation at 5000 g at 4°C for 10 minutes, the supernatants were immunoprecipited with 35 µl of anti HA-agarose beads (Sigma). These were washed five times with 500 µl of PBS supplemented with 1% triton-X 100, followed by a final wash with 500 µl of kinase buffer (Cell Signaling) and re-suspension in 30 µl of kinase buffer. To this was added 1 µl of ATP labelled with 32P at the γ phosphate position to a final concentration of 50 nM (specific activity 3000 Ci/mmol) (Perkin Elmer). Assays were incubated for 1 hour at 30°C with shaking. The assay was terminated by denaturation in 4x sample buffer (Invitrogen) plus 5% β-Mercaptoethanol (Sigma) and run on 4-12% Bis Tris gels (Invitrogen). Following transfer onto PVDF membrane, the membranes were exposed to a phosphorscreen for 48 hours and scanned using a STORM 840 phosphorimager (GE Healthcare). Membranes were subsequently probed for total protein as described above. Band intensities were quantified using ImageJ (NIH) and the kinase activity for each construct corrected for total protein.
Immunocytochemistry (ICC)
For immunocytochemistry HEK293T cells were grown on 5 μg/ml poly-D-lysine (Sigma) coated coverslips and fixed with 4% PFA at room temperature for 10 minutes. Fixed cells were blocked with 15% normal goat serum (NGS) in PBS-T (PBS containing 0.1% Triton-X) prior to incubation with primary antibodies in 10% NGS PBS-T (1:2000 rat anti-HA, Roche; 1:1000 mouse anti-LAMP1, Abcam). Next cells were incubated with fluorescently labeled secondary antibodies in 10% NGS in PBS-T (goat anti rat 488, goat anti mouse IgG1 568 respectively) before being counterstained with DAPI at 1:2000 in PBS and mounted onto glass slides (VWR) using Fluoromount G mounting medium (SouthernBiotech). F-Actin was detected by staining with 1:300 Alex fluor 633 phalloidin (Life Technologies). Finally images were obtained using a LSM 710 confocal microscope and images were compiled using Adobe Photoshop.
Blue Native Gel Electrophoresis
Blue native gel electrophoresis was carried out using the Native PAGE system (Life technologies) as per manufacturers instructions. Confluent 10 cm diameter plates of HEK293T cells transfected with wild type, K42W and T701N DAPK1 constructs were harvested and lysed in 5% digitonin. 20 μl of each lysate was loaded onto 3-12% Blue NativePAGE gels and run at 150V for 110 minutes. Gels were transferred to PVDF membrane as described above with the addition of 0.1% SDS to the transfer buffer.
Size Exclusion Chromatography (SEC)
Transiently transfected HEK293T were harvested in LB and cleared lysates (0.5 ml) were injected and separated on a Superose 6 10/300 column (GE Healthcare). The column was per-equilibrated with buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.07% Tween 20) and used at a flow rate of 0.5 ml/min. Elution volumes of standards were 7.5 ml for Blue Dexatran (V0), 11.5 ml for hemocianin from Carcinus aestuarii (900 kDa), 12 ml for thyreoglobin (669 kDa), 14 ml for ferritin (440 kDa), 15.5 ml for catalase (232 kDa) and 17 ml for aldolase (158 kDa). Elution fractions were analyzed by dot blot (see below).
Dot Blot
One microliter of each fraction from SEC was applied onto a nitrocellulose membrane. The membrane was blocked with 5% milk for 1 hour and subsequently incubated with rat monoclonal anti-HA clone 3F10 (Roche) followed by secondary anti-rat IgG (Sigma) in 5% (w/v) milk.
Transmission electron microscopy
HA-DAPK1 positive fractions collected after SEC were combined and diluted 20 times. Aliquots of HA-DAPK1 enriched samples were adsorbed to a glow-discharged carbon-coated copper grid and subsequently gold-labelled with mouse monoclonal anti-HA for one hour. After washing, samples were incubated with 5 nm gold-labelled mouse anti IgG secondary antibody for one hour. Negatively stained samples were prepared using previously described methods47. Each gold labelled grid was washed with two drops of deionized water and stained with two drops of freshly prepared 1% uranyl acetate. Samples were imaged at room temperature using a Fei Tecnai T12 electron microscope equipped with a LaB6 filament and operated at an acceleration voltage of 100 kV. Images were taken at calibrated magnifications in the range 11,000 x to 26,000 x.
The distribution of distances among all immunogold labelled proteins (n = 559) was obtained using ImageJ (NIH, Bethesda, MD, USA). Distances greater than 100 nm were discarded from the analysis. Frequency distribution of particles distances was carried out using graph Prism setting 5 nm as bin size. The obtained data set was normalized by the annulus area within which the particles were counted (thickness corresponds to the bin size, 5 nm):

nd represents the number of gold-labelled particles counted inside the annulus area (2πr2d) – (2πr2d-(d-5)) and d is a given distance from the analyzed particles.
Statistics
Statistical comparisons were carried out using the graph prism suite of software. One-way analysis of variance analysis (ANOVA) was carried out with Tukey’s post hoc correction. Significance was considered to be <0.05.
References
Bosgraaf, L. & Van Haastert, P. J. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta 1643, 5-10 (2003).
Lewis, P. A. The function of ROCO proteins in health and disease. Biol Cell 101, 183-191 (2009).
Dachsel, J. C. & Farrer, M. J. LRRK2 and Parkinson disease. Arch Neurol 67, 542-547 (2010).
Simon-Sanchez, J. et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 41, 1308-1312 (2009).
Barrett, J. C. et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 40, 955-962 (2008).
Zhang, F. R. et al. Genomewide association study of leprosy. N Engl J Med 361, 2609-2618 (2009).
Inbal, B. et al. DAP kinase links the control of apoptosis to metastasis. Nature 390, 180-184 (1997).
Raveh, T., Droguett, G., Horwitz, M. S., DePinho, R. A. & Kimchi, A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol 3, 1-7 (2001).
Raval, A. et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 129, 879-890 (2007).
Gozuacik, D. & Kimchi, A. DAPk protein family and cancer. Autophagy 2, 74-79 (2006).
Bialik, S. & Kimchi, A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin Cancer Biol 14, 283-294 (2004).
Li, Y. et al. DAPK1 variants are associated with Alzheimer's disease and allele-specific expression. Hum Mol Genet 15, 2560-2568 (2006).
Minster, R. L., DeKosky, S. T. & Kamboh, M. I. No association of DAPK1 and ABCA2 SNPs on chromosome 9 with Alzheimer's disease. Neurobiol Aging 30, 1890-1891 (2009).
Dachsel, J. C. et al. Death-associated protein kinase 1 variation and Parkinson's disease. European journal of neurology : the official journal of the European Federation of Neurological Societies 18, 1090-1093 (2011).
Deng, J. et al. Structure of the ROC domain from the Parkinson's disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc Natl Acad Sci U S A 105, 1499-1504 (2008).
Gotthardt, K., Weyand, M., Kortholt, A., Van Haastert, P. J. & Wittinghofer, A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. Embo J 27, 2239-2249 (2008).
Tereshko, V., Teplova, M., Brunzelle, J., Watterson, D. M. & Egli, M. Crystal structures of the catalytic domain of human protein kinase associated with apoptosis and tumor suppression. Nature structural biology 8, 899-907 (2001).
Gilsbach, B. K. et al. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proceedings of the National Academy of Sciences of the United States of America 109, 10322-10327 (2012).
Ito, G. et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry 46, 1380-1388 (2007).
Korr, D. et al. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal 18, 910-920 (2006).
Weiss, B. ROCO kinase activity is controlled by internal GTPase function. Science signaling 1, pe27 (2008).
Cohen, O., Feinstein, E. & Kimchi, A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. Embo J 16, 998-1008 (1997).
Smith, W. W. et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9, 1231-1233 (2006).
Lee, B. D. et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med 16, 998-1000 (2010).
Greggio, E. et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23, 329-341 (2006).
Deng, X. et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 7, 203-205 (2011).
Okamoto, M. et al. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J Med Chem 52, 7323-7327 (2009).
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912-1934 (2002).
Marin, I. The Parkinson disease gene LRRK2: evolutionary and structural insights. Molecular biology and evolution 23, 2423-2433 (2006).
Carlessi, R. et al. GTP binding to the ROC domain of DAP-kinase regulates its function through intramolecular signalling. EMBO reports, 9, 917-923 (2011).
Lewis, P. A. et al. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun 357, 668-671 (2007).
West, A. B. et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 16, 223-232 (2007).
Daniels, V. et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. Journal of neurochemistry 116, 304-315 (2011).
Taymans, J. M. et al. LRRK2 Kinase Activity Is Dependent on LRRK2 GTP Binding Capacity but Independent of LRRK2 GTP Binding. PLoS One 6, e23207 (2011).
Jin, Y., Blue, E. K. & Gallagher, P. J. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. The Journal of biological chemistry 281, 39033-39040 (2006).
Webber, P. J. et al. Autophosphorylation in the Leucine-Rich Repeat Kinase 2 (LRRK2) GTPase Domain Modifies Kinase and GTP-Binding Activities. Journal of molecular biology 412, 94-110 (2011).
Rudenko, I. N. et al. The G2385R Variant of Leucine-Rich Repeat Kinase 2 Associated with Parkinson's Disease is a Partial Loss of Function Mutation. The Biochemical journal, 446, 99-111 (2012).
Greggio, E. et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem 283, 16906-16914 (2008).
Sen, S., Webber, P. J. & West, A. B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem 284, 36346-36356 (2009).
Gasper, R., Meyer, S., Gotthardt, K., Sirajuddin, M. & Wittinghofer, A. It takes two to tango: regulation of G proteins by dimerization. Nat Rev Mol Cell Biol 10, 423-429 (2009).
Berger, Z., Smith, K. A. & Lavoie, M. J. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511-5523 (2010).
Civiero, L. et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS One 7, e43472 (2012).
Ito, G. & Iwatsubo, T. Re-examination of the dimerization state of leucine-rich repeat kinase 2: predominance of the monomeric form. The Biochemical journal, 441, 987–994 (2011).
Klein, C. L. et al. Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J Neurochem 111, 703-715 (2009).
Monod, J., Wyman, J. & Changeux, J. P. On the Nature of Allosteric Transitions: A Plausible Model. Journal of molecular biology 12, 88-118 (1965).
Monod, J., Changeux, J. P. & Jacob, F. Allosteric proteins and cellular control systems. Journal of molecular biology 6, 306-329 (1963).
Ohi, M., Li, Y., Cheng, Y. & Walz, T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online 6, 23-34 (2004).
Acknowledgements
The authors would like to thanks Dr Selina Wray and Professor Luigi Bubacco for critical comments. This work was supported in part by the Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson's Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield and the MRC Protein Phosphorylation Unit at the University of Dundee, by grants from the Michael J. Fox foundation for Parkinson’s research to P.A.L. and E.G and MIUR (Rientro dei Cervelli, lncentivazione alla mobilità di studiosi stranieri e italiani residenti all’estero) to E.G. P.A.L. is a Parkinson’s UK research fellow (grant F1002). This work was supported in part by funds from University College London graduate school (J.D.J.).
Author information
Authors and Affiliations
Contributions
Experiments were planned by E.G. and P.A.L. Experiments were carried out by J.J., S.D., L.C., C.M., E.G. and P.A.L. The manuscript was written by E.G. and P.A.L. and was viewed and commented on by all the authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Jebelli, J., Dihanich, S., Civiero, L. et al. GTP binding and intramolecular regulation by the ROC domain of Death Associated Protein Kinase 1. Sci Rep 2, 695 (2012). https://doi.org/10.1038/srep00695
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00695
This article is cited by
-
Role and mechanism of action of leucine-rich repeat kinase 1 in bone
Bone Research (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
