
Abstract
Anaplasma marginale infection is one of the most common tick-borne diseases, causing a substantial loss in the beef and dairy production industries. Once infected, the pathogen remains in the cattle for life, allowing the parasites to spread to healthy animals. Since clinical manifestations of anaplasmosis occur late in the disease, a sensitive, accurate, and affordable pathogen identification is crucial in preventing and controlling the infection. To this end, we developed an RPA-CRISPR/Cas12a assay specific to A. marginale infection in bovines targeting the msp4 gene. Our assay is performed at one moderately high temperature, producing fluorescent signals or positive readout of a lateral flow dipstick, which is as sensitive as conventional PCR-based DNA amplification. This RPA-CRISPR/Cas12a assay can detect as few as 4 copies/μl of Anaplasma using msp4 marker without cross-reactivity to other common bovine pathogens. Lyophilized components of the assay can be stored at room temperature for an extended period, indicating its potential for field diagnosis and low-resource settings of anaplasmosis in bovines.
Similar content being viewed by others

Introduction
Anaplasmosis is an infectious blood disease distributed globally across five continents caused by Anaplasma marginale, an obligate intracellular parasitic bacterium1,2,3,4,5,6,7,8. The parasite is carried by blood-feeding insects such as ticks and flies, and bovines can contract the disease through insect bites or direct contact with traces of blood from infected animals9. The disease can be treated with extensive administration of tetracyclines although permanent clearance of the parasites remains questionable10,11,12.
Clinical manifestations of A. marginale infection are marked by acute onset fever and other nonspecific symptoms such as decreased appetite and general weakness, similar to other insect-borne infectious blood diseases, such as, babesiosis caused by Babesia spp. and trypanosomiasis caused by Trypanosoma spp13,14,15. Hence, A. marginale infection is often misdiagnosed or diagnosed late in the disease state, and can only be confirmed by the identification of the infectious bacterium.
Conventionally, A. marginale infection is determined by microscopic observation, which is inherently subjective, requiring an experienced veterinary pathologist capable of discerning A. marginale from other bacterial parasites, and is prone to produce false negatives in cases of low levels of the pathogen16. Serological tests such as latex agglutination, complement fixation, indirect fluorescent antibody, and enzyme-linked immunosorbent assay (ELISA) identify the parasite based on the detection of IgG antibodies against A. marginale antigens, including major surface protein 2 (MSP2), MSP4, and MSP516. These tests are considered simple, and affordable, providing sufficient accuracy for clinical diagnosis17,18; however, cross-reactivity of the chosen antibody can affect the specificity of the test16,18.
Molecular detection has been employed in the identification of A. marginale in cattle blood samples. Polymerase chain reaction (PCR)-based methods or their modifications have been used to amplify a selected region of the A. marginale genome, followed by visualization with gel electrophoresis methods. While identification based on PCR-based DNA amplification is typically more sensitive and specific, it is considered time-consuming and less applicable to fieldwork due to the storage requirement of reagents at low temperatures and specific laboratory instruments such as a thermocycler and high thermal power16,19,20. To overcome challenges in point-of-care diagnostics, recombinase polymerase amplification (RPA), a form of isothermal amplification, and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas12a have been combined and applied for the detection without compromising the sensitivity or specificity of molecular detection. In RPA-CRISPR/Cas12a, amplicon specificity is determined by the cleavage of the endonuclease Cas12a, which is guided by a specific CRISPR RNA (crRNA) that recognizes the target sequence21,22. The RPA-CRISPR/Cas12 system and the modifications have been successfully developed for one-tube, point-of-care testing of African swine fever, SARS-CoV2, and monkeypox, displaying enhanced sensitivity and accuracy suitable for point-of-care diagnosis23,24,25,26,27.
In this study, the RPA-CRISPR/Cas12a system was developed for the diagnosis of A. marginale infection in bovines, which, to our knowledge, has not been previously reported. The readouts of our assay can be in fluorescent or colorimetric lateral flow dipstick formats, and the specificity is on par with PCR-based amplification. Also, the test reagents of our assay can be stored at room temperature, making it suitable for on-site diagnosis in low-resource settings, providing an optional on-site diagnostic method that can significantly improve the containment and management of A. marginale infection in livestock.
Results
In our assay, cattle blood samples were collected, and the genomic DNA was extracted for msp4 amplification by RPA. Following RPA, specific CRISPR RNA (crRNA) was used to guide the Cas12a endonuclease to cleave the RPA amplicons, where positive detection can be translated into fluorescent signals or a positive read-out of lateral-flow dipsticks (LFD) (Fig. 1).

The RPA-CRISPR/Cas12a assay for the diagnosis of Anaplasma marginale infection in bovines comprises blood sample collection, genomic DNA extraction, DNA amplification by RPA, and detection by CRISPR/Cas12a. Positive readout (P) can be translated into fluorescent signals or a single-color band in a lateral-flow dipstick (LFD), while negative readout (N) results in the absence of fluorescent signal or two-color bands in LFD.
Development of the RPA-CRISPR/Cas12a assay
RPA using F1-R1 or F2-R1 as primers resulted in 269 bp and 219 bp amplified fragments, respectively, and the combination of all three primers (F1-F2-R1) produced both of the amplified fragments (Fig. 2B). The RPA products from all primer combinations were subsequently detected by CRISPR/Cas12a using an equal molar amount of the crRNA. The three-primer combination displayed the most intense fluorescence, followed by F1-R1 and F2-R1 combinations, respectively (Fig. 2C,D). For this reason, the three-primer combination was chosen for our RPA-CRISPR/Cas12a assay.

Validation of RPA-CRISPR/Cas12a assay detection. (A) The target sequence of the A. marginale msp4 gene region. RPA primers consist of 2 forward (F1 and F2) and 1 reverse (R1) direction as shown in red were designed to flank the PAM sequence (bold letters) and the crRNA sequence (underlined). (B–D) Primer combination for assay condition optimization. The agarose gel of RPA products and corresponding fluorescent signals of CRISPR/Cas12a detection were shown, respectively. The bar graph represents the data of mean ± SD from 3 independent experiments. One-way ANOVA with a Tukey's multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. (E) Limit of detection (LOD) test. Gel electrophoresis showing RPA amplification products of serially diluted msp4 recombinant plasmid. The starting concentration of the plasmid template is indicated below each lane. N is no template control. L is 50 bp DNA ladder (ExcelBand™). (F,G) Fluorescent signals of CRISPR/Cas12a reactions from 1 μl of RPA products in (E). The fluorescence intensities were quantitated by ImageJ and plotted by GraphPad Prism version 6 (GraphPad software, Boston, USA). The data represents as mean ± SD from 9 independent experiments. One-way ANOVA with a Tukey's multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 versus control.
Limit of detection (LOD)
To determine the limit of detection of our Anaplasma diagnosis by RPA-CRISPR/Cas12a fluorescent assay, msp4 plasmid was used as a DNA template in a tenfold serial dilution covering a range from 3 pg to 3 ag. Only one-fifth of RPA amplification product was subsequently applied to CRISPR/Cas12a assay and the rest was loaded onto agarose gel electrophoresis to assess amplification efficiency (Fig. 2E). Fluorescent readout of the end product from RPA-CRISPR/Cas12 assay demonstrates the assay could distinctly detect as low as 30 ag/μl of the starting DNA material (Fig. 2F,G), equivalent to 4 copies/μl of Anaplasma msp4 as calculated from the size of plasmid DNA template. LOD for this assay, in comparison to the PCR method, was additionally investigated by applying % hit rate plots to determine copies of starting DNA material required at 95% confidence27. The same set of primers were used for both assays. However, only one 219-bp DNA product band was visually observed by PCR method despite intensive optimizations. At 95% confidence, data suggest the two assays have equivalent LOD of 21 and 20 copies/µl for RPA and PCR amplification, respectively (Supplementary Table 1, Supplementary Fig. 1).
Cross-reactivity test
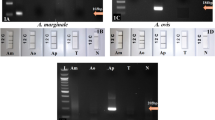
To evaluate the detection of A. marginale infection and the extent of cross-reactivity, we challenged the assay with genomic DNA extracted from blood samples of bovines infected with other vector-borne diseases. The infecting pathogen was confirmed by PCR-based DNA amplification targeting the msp5 gene of A. marginale, sbp2 (spherical body protein 2) of B. bovis, p23 of T. orientalis, and the 18S region of T. evansi (Fig. 3A) before the samples were used as a template for the RPA-CRISPR/Cas12a assay. As a result, only the samples with confirmed A. marginale infection displayed visible fluorescent signals, establishing that our RPA-CRISPR/Cas12a assay is specific to A. marginale infection (Fig. 3B).

Cross-reactivity evaluation. (A) Gel electrophoresis showing PCR amplification of genomic DNA extracted from cattle blood infected with A. marginale, B. bovis, T. orientalis, or T. evansi (L: 100 bp DNA ladder (Solis BioDyne, Estonia). The target genes and their primer sequences are listed in Table 1. (B) The agarose gel image of RPA reaction and the fluorescent signals of CRISPR/Cas12a reactions using genomic DNA extracted from cattle blood infected with A. marginale, B. bovis, T. orientalis, or T. evansi (N: no template control). (L: 50 bp DNA ladder (ExcelBand™).
Assay performance
The accuracy of our RPA-CRISPR/Cas12a was evaluated using the genomic DNA extracted from 100 cattle blood samples. PCR-based DNA amplification targeting the msp4 gene established that half of the blood samples were confirmed positive and half were negative for A. marginale infection (Supplementary Fig. 2). When using the same amount of DNA template (3 ng) in our assay, all 50 samples that were confirmed negative by PCR tested negative, 46 out of 50 samples that were confirmed positive by PCR tested positive, and 4 out of 50 produced false negatives (Supplementary Table 2, Supplementary Fig. 3). Accordingly, the assay's sensitivity, specificity, and accuracy were 92, 100, and 96%, respectively (MedCalc Software Ltd. Diagnostic test evaluation calculator; Version 22.009).
Stability and storability test
Resuspended lyophilized reactions produced true negative and positive results regardless of the storage temperature (Fig. 4A). The fluorescent signals from lyophilized reactions stored at 25 and 37 °C were lower than those stored at − 20 or 4 °C. Nonetheless, the fluorescence emanating from positive samples is distinct from negative samples so that it can be correctly interpreted, indicating that the assay is stable and stored at room temperature. Next, we extended the storage time at 37 °C from 1 week to up to 5 weeks. At the first four time points, the resuspended lyophilized reactions could produce true negatives and positives, suggesting its capability for long-term storage (Fig. 5B,C).

Stability and storability test. (A) The RPA-CRISPR/Cas12a results from resuspended lyophilized RPA and CRISPR/Cas12a components, which were stored at different temperatures for 1 week. (B,C) The RPA-CRISPR/Cas12a results from resuspended lyophilized RPA and CRISPR/Cas12a components, which were stored at 37 °C from 1 week up to 5 weeks. The average intensities of the fluorescent signals were quantitated and represented as mean ± SD. n = 3.

Schematic representation of the lateral flow dipstick chromatography (LFD) and visual readouts of RPA-CRISPR/Cas12a reaction. The white arrows on the dipsticks indicate the end of the stick to dip. The following templates are shown: (−) plasmid DNA, (+) msp4 recombinant plasmid DNA, (P1–P3) genomic DNA extracted from blood drawn from bovines known to be infected with A. marginale, and (N1–N3) genomic DNA extracted from blood drawn from bovines free from A. marginale infection.
Lateral flow dipsticks
As fluorescent signal detection requires a specialized transilluminator, we adapted the assay readout to lateral-flow dipsticks (LFD). The RPA-CRISPR/Cas12a reaction solutions were applied at the conjugate pad of the LFD. For a negative sample, where the fluorescein (FITC)-biotin probes are not cleaved by Cas12a enzyme, the intact probes will interact with the gold nanoparticle (AuNP) at the conjugate pad and then bind to the anti-BIOTIN capture antibody. An excess solution containing an uncleaved probe will travel forward and bind to an anti-mouse IgG antibody, resulting in the appearance of two bands on a strip. In contrast, the positive sample appears as a single band as the probes have been cleaved. The FITC side of the probe will bind to the anti-FITC capture antibody embedded on the AuNP and thereby be trapped by the anti-mouse IgG antibody (Fig. 5). The presence of AuNP generates the signal through surface plasmon resonance of the gold nanoparticle, which is observed as a red fluorescence. The cleaved probe containing the biotin will still bind to the anti-biotin antibody, however the AuNP is lost after cleavage and no signal will be observed. The resulting reaction of msp4 recombinant plasmid and genomic DNA extracted from samples positive for A. marginale infection appeared as one colored band, whereas genomic DNA obtained from samples negative for A. marginale infection resulted in two colored bands (Fig. 5). Thus, LFD can be used as the readout of our RPA-CRISPR/Cas12a assay instead of fluorescent signals.
Discussion
The uncertainty in diagnosis of the disease also has major public health concerns and economic impact on farmers. In Thailand’s rural areas, several tick-borne diseases exist28. Livestock husbandry, especially cattle, are fed by wandering and grazing natural bush in open areas where sanitation is poorly controlled. Thus, cattle are at high risk of infection, resulting in substantial losses to rural revenue29. Various epidemiological surveys of tick-borne pathogen species have been performed in different parts of Thailand. The infections were confirmed by PCR molecular detection methods largely focusing on the msp (major surface protein) gene for Anaplasmataceae family3,30. Over 65% of infection rates were identified as A. marginale from positive blood samples collected from 18 provinces over 5 different regions of Thailand31. However, the prevalence of the disease varies due to different seasons and times, even though the blood samples were collected from the same region of the country32,33.
Several diagnostic methods for A. marginale have been well established34. However, the optimum diagnosis depends on the detection time relative to the time of infection and species identifiability from testing. This study reports a new nucleic acid detection method for A. marginale pathogen in infected cattle. The major surface protein 4 gene (msp4) was selected as a gene target. MSP4 is one of six members that belong to the major surface protein (MSP) superfamily. These proteins play a crucial role in the adhesion of A. marginale to host cells’ erythrocytes causing the infection11. MSPs are thus the most likely gene candidates selected for Anaplasma spp. for molecular detection. Particularly msp4 and msp5 that are shown to be highly conserved because they have originated from single genes35,36. Besides msp, other genes such as ankA, groESL and 16S rRNA, are occasionally selected as target genes37. The 16S rRNA gene is also widely used as a marker for bacterial identification. Nonetheless, the efficiency of detection would also depend on the gene regions that are amplified. Some areas of the gene are more conserved compared to other regions of the same gene38. Our detection assay comprises two steps after genomic DNA preparation: specific gene amplification and signal detection. For the first step, the gene target is amplified by isothermal nucleic acid amplification, recombinase polymerase amplification (RPA)39. RPA has revolutionized the isothermal amplification technology and has been used to detect a wide range of pathogens in human and veterinary medicine, agriculture, and food safety40,41,42,43. Similar to conventional PCR, a pair of primers is needed to amplify the target spanning the amplicons that are not longer than 300 bp for the most efficient amplifiability. To obtain more amplicon products, we employed two forward primers (F1 and F2) to pair with one reverse primer (R1), yielding two amplified fragments. This strategy has drastically increased the amplified products and the assay's sensitivity for the final detection. Both amplified products contain the protospacer adjacent motif (PAM) required for the CRISPR/Cas12a detection system in the second step. PAM sequence is a short DNA motif required for the Cas endonuclease enzyme to recognize and then cleave on the DNA target. Initially, the Cas12a enzyme forms a complex with CRISPR RNA (crRNA), guides RNA for sequence-specific targets, and then starts searching for the PAM recognition sequence (TTTV). Once the PAM sequence is found, the crRNA binds to the complementary sequence of the target DNA adjacent to the PAM sequence. The enzyme now undergoes major conformational changes that activate catalytic activity, followed by cleavage of the double-stranded target DNA. After cleavage, the Cas12a enzyme remains activated, exhibiting a collateral cleavage activity where the enzyme can cleave the surrounding single-stranded DNA (ssDNA) non-specifically44,45,46. CRISPR/Cas12a detection assay contains a reporter probe consisting of a quencher and a fluorophore with a short ssDNA. Consequently, the activated Cas12a enzyme collaterally cleaves the nearby reporter probe at the ssDNA, separating the quencher and fluorophore, allowing the fluorescence to be detected47. The fluorescence intensity represents the amount of DNA that has been amplified, which correlates with the ability of the crRNA to bind to the target DNA in a complementary manner. The performance of the assay is determined by a limit of detection (LOD). This study demonstrated that the RPA-CRISPR/Cas12a assay targeting msp4 for A. marginale detection is excellent. We can detect the starting DNA material down to 30 ag, which is equal to 4 copies/μl of msp4 per reaction. The clinical validation was also performed with cattle blood samples. The diagnostic performance in sensitivity, specificity, and accuracy are met. A cross-reaction with other pathogens causing similar clinical symptoms to anaplasmosis was not observed. The fluorescent signal of the detection can be visualized by a portable device. The lateral flow assay format can also be conveniently deployed by changing reporter probes labeled with reporter molecules corresponding with the antibodies bound to the strip.
Initially, both of these detection formats offer a simplified approach to nucleic acid detection with some technical limitations. This was due to several components of the assay reaction, such as fluorescent probes and enzymes, requiring an ultra-low temperature to protect them from degradation and denaturation. Therefore, employing this detection kit would be difficult at a point of care, especially in rural areas where the cold chain transportation and electricity could not be accessed. Several studies have attempted to prolong the stability of the reaction components at ambient temperature48,49,50. In this study, we have overcome this issue by adding specific stabilizers to preserve the structure of the biological molecules. The lyophilized reactions remain stable for up to a month, even stored at high temperatures (37 °C), thereby maintaining the activity of the protected components. Moreover, the assay operation has been simplified by putting all reaction components in one tube for each reaction step, resulting in a highly convenient detection kit for the end user.
Conclusion
The RPA-CRISPR/Cas12a assay developed in this study provides an alternative to identifying A. marginale by DNA amplification. The specificity and accuracy of our assay are on par with conventional PCR-based assays. The assay can be performed in under an hour at a moderately high temperature (39 °C), achievable with a conventional incubator, heat block, or lukewarm water bath. The lyophilization of the assay components and readout by LFD also demonstrate its potential for long-term storage and on-site field diagnosis of anaplasmosis in bovines.
Materials and methods
Study areas and sample collection
The cattle blood samples in this work were obtained from previous studies32,51. They are collected from different areas in the northern and northeastern parts of Thailand. Briefly, blood samples were collected from the jugular vein of cattle, which were chosen randomly regardless of their clinical symptoms. Fifty A. maginale-infected and non-infected samples were selected for this study. All study protocols were approved by the institutional committee for the use and care of laboratory animals (Ethical Approval Protocol No. IMB-ACUC 2023/008) and were performed in accordance with the relevant guidelines and regulations. These studies were also reported as described by the ARRIVE guidelines 2.0. Once collected, the sample is transferred to an individual tube containing ethylenediaminetetraacetic acid (EDTA) buffer solution and kept at 4 °C.
Nucleic acid preparation
A recombinant plasmid containing the A. marginale msp4 gene obtained from previous study36 was used as a DNA template for positive controls in PCR and RPA. The recombinant plasmid was transformed and maintained in Escherichia coli DH5α, cultured overnight, and the plasmid was extracted using the Large Plasmid DNA Extraction Kit (Geneaid Biotech Ltd.). The plasmid was verified based on the insert size using restriction digestion and agarose gel electrophoresis.
Genomic DNA was extracted from 200 μl of whole blood by a High Pure PCR Template Preparation kit (Roche) following the manufacturer's protocol and kept at − 20 °C until used.
Examination of parasitic infections and PCR-based DNA amplification
Collected blood samples were analyzed for infection with A. marginale, Babesia bovis, Theileria orientalis, and Trypanosoma evansi by PCR-based DNA amplification using the extracted genomic DNA. Each PCR reaction contained 1 μM of forward and reverse primers, 200 μM of each dNTP, 1× Phusion™ HF buffer, 0.5 U Phusion DNA polymerase (Thermo Scientific™, F-530S), and 50 ng of the template DNA unless indicated otherwise (Supplementary Table 3). PCR reactions were performed using the following condition: 94 °C for 5 min, 30 cycles of 94 °C for 1 min, 55 °C for 1 min, 68 °C for 1 min, and 68 °C for 7 min. The PCR products were separated by 1% agarose gel electrophoresis and stained with 0.1% ethidium bromide solution. The target gene for each pathogen and the sequence of forward and reverse primers, including the expected length of the PCR amplicon(s), are listed in Table 1.
DNA amplification by recombinase polymerase amplification (RPA)
For the DNA amplification step, three primers were designed to target the msp4 gene of A. marginale. The combination of the reverse primer R1 and the forward primer F1 or F2 flanks the nucleotides of the protospacer adjacent motif (PAM) sequence (5ʹ-TTTC-3ʹ) and the crRNA sequence (5ʹ-CTTCTGTTACCTCGTTCGA-3ʹ), which are required for the downstream CRISPR/Cas12a detection (Fig. 2A).
DNA amplification by RPA was performed using TwistAmp™ Basic Kit (TwistDX). Either recombinant DNA or genomic DNA was used as a template. Briefly, 0.3 μM of each forward primer, 0.6 μM of the reverse primer targeting the A. marginale msp4 gene were added to 29.5 μl of rehydration buffer to resuspend lyophilized components, and 14 mM of magnesium acetate was added just before the reaction commenced to a final volume of 50 μl (Supplementary Table 3). RPA was performed at 39 °C for 15–30 min, and the amplification was stopped by incubating at 75 °C for 5 min. The RPA products were preliminarily verified by 2% (w/v) agarose gel electrophoresis before detection by CRISPR/Cas12a. The sequence of forward and reverse primers, including the expected length of the RPA amplicon, are listed in Table 1.
Cas12a expression and purification
The Cas12a protein was expressed and purified from E. coli BL21, harboring the Cas12a plasmid (Addgene plasmid #114366). The expression, purification, and quantification of Cas12a were performed as previously described52. The purity of Cas12a was visualized on 10% SDS-PAGE gel, stored in 50% glycerol, and kept at − 20 °C until used.
CRISPR/Cas12a detection
The RPA products were detected in a CRISPR/Cas12a reaction containing 1 μl of RPA product, 30 nM crRNA, 1X NEBuffer 2.0, 50 nM Cas12a enzyme, and 1.5 μM of the fluorescent reporter of single strand DNA probe with fluorescein (FAM) and black hole quencher 1 (BHQ1) at the 5ʹ and 3ʹ ends, respectively (Supplementary Table 3). The reaction was incubated at 39 °C for 15–30 min, and the resulting fluorescence was visualized by eyes using BluPAD Dual LED Blue/White Light Transilluminator (Bio-helix, BP001CU). For detection by lateral flow dipsticks (LFD) with the use of Amplicon detection 1T1C dip strip (Serve Science Co. Ltd., Thailand), the reaction was performed in the same manner but with a fluorescein (FITC)-biotin probe instead of FAM-quencher probe.
Limit of detection (LOD)
The developed assay's limit of detection (LOD) was evaluated using the msp4 recombinant plasmid. The starting concentration of the DNA template was determined using the NanoDrop 2000™ spectrophotometer (Thermo Scientific), and 3 pg/μl of the plasmid DNA was serially diluted tenfold until the final concentration reached 3 ag/μl. For RPA-CRISPR/Cas12a, the images were used to quantify the fluorescence intensity by ImageJ53 and the values from independent RPA-CRISPR/Cas12a experiments were averaged, analyzed, and plotted by GraphPad Prism version 6 (GraphPad software, Boston, USA.).
Storage and stability of assay components
The assay components were tested for storage stability after freeze-drying. To increase the stability and storability of the assay, we used a combination of cryoprotectants and freeze-dried all components to preserve the enzymes and crRNA used in the assay. Cryoprotectants were added to RPA and CRISPR/Cas12a components before lyophilization. For RPA, 10% trehalose, 5% polyethylene glycol 35 (PEG35), and 0.1% Triton X-100 were used, and 10% trehalose, 5% pullulan, and 0.1% Triton X-100 were for CRISPR/Cas12a. Lyophilization was performed separately using a Flexi-Dry™ MP freeze-dryer (Kinetics, USA) for 1 h or until the reactions were wholly dried, as previously described 52. To test the storage stability of assay components at different temperatures, the lyophilized reactions were stored at − 20, 4, 25, or 37 °C for 1 week and tested with genomic DNA extracted from blood samples that tested positive for A. marginale infection. The stability of freeze-dried components at 37 °C were assessed by storing the lyophilized reagents at 37 °C for 1, 2, 3, and 4 weeks. After each designated storage time, the lyophilized reagents were kept at − 30 °C until all reactions could be resuspended and tested simultaneously.
Ethical approval and consent to participate
Blood sample collection protocol, including sampling number and the handling of animals, was approved by the Institutional Animal Care and Use Committee (IACUC), Institute of Molecular Biosciences, Mahidol University (Protocol No. IMB-ACUC 2023/008), with consent to collect blood samples from owners at the collection site.
Data availability
All data presented in this study are available in the article.
References
El-Alfy, E. S. et al. Global prevalence and species diversity of tick-borne pathogens in buffaloes worldwide: A systematic review and meta-analysis. Parasit. Vectors 16, 115. https://doi.org/10.1186/s13071-023-05727-y (2023).
Zhyldyz, A. et al. Epidemiological survey of Anaplasma marginale in cattle and buffalo in Sri Lanka. J. Vet. Med. Sci. 81, 1601–1605. https://doi.org/10.1292/jvms.19-0242 (2019).
Seerintra, T., Saraphol, B., Thanchomnang, T. & Piratae, S. Molecular prevalence of Anaplasma spp. in cattle and assessment of associated risk factors in Northeast Thailand. Vet. World 20, 1702–1707. https://doi.org/10.14202/vetworld.2023.1702-1707 (2023).
Fathi, A. et al. Molecular identification, risk factor assessment, and phylogenetic analysis of tick-borne pathogens in symptomatic and asymptomatic cattle from South-Eastern Iran. Exp. Appl. Acarol. https://doi.org/10.1007/s10493-023-00886-0 (2024).
Segura, J. A. et al. Molecular surveillance of microbial agents from cattle-attached and questing ticks from livestock agroecosystems of Antioquia, Colombia. Comp. Immunol. Microbiol. Infect. Dis. https://doi.org/10.1016/j.cimid.2023.102113 (2024).
Sisson, D., Beechler, B., Jabbar, A., Jolles, A. & Hufschmid, J. Epidemiology of Anaplasma marginale and Anaplasma centrale infections in African buffalo (Syncerus caffer) from Kruger National Park, South Africa. Int. J. Parasitol. Parasites Wildl. 21, 47–54. https://doi.org/10.1016/j.ijppaw.2023.04.005 (2023).
Moraga Fernandez, A. et al. Fatal cases of bovine anaplasmosis in a herd infected with different Anaplasma marginale genotypes in southern Spain. Ticks Tick Borne Dis. 13, 101864. https://doi.org/10.1016/j.ttbdis.2021.101864 (2022).
Gofton, A. W. et al. Detection and phylogenetic characterisation of novel Anaplasma and Ehrlichia species in Amblyomma triguttatum subsp. from four allopatric populations in Australia. Ticks Tick Borne Dis. 8, 749–756. https://doi.org/10.1016/j.ttbdis.2017.05.009 (2017).
Jaswal, H., Bal, M. S., Singla, L. D., Gupta, K. & Brar, A. P. Pathological observations on clinical Anaplasma marginale infection in cattle. J. Parasit. Dis. 39, 495–498. https://doi.org/10.1007/s12639-013-0384-4 (2015).
Sarli, M. et al. Efficacy of long-acting oxytetracycline and imidocarb dipropionate for the chemosterilization of Anaplasma marginale in experimentally infected carrier cattle in Argentina. Vet. Parasitol. Reg. Stud. Rep. 23, 100513. https://doi.org/10.1016/j.vprsr.2020.100513 (2021).
Aubry, P. & Geale, D. W. A review of bovine anaplasmosis. Transbound. Emerg. Dis. 58, 1–30. https://doi.org/10.1111/j.1865-1682.2010.01173.x (2011).
Curtis, A. K. et al. Failure to eliminate persistent Anaplasma marginale infection from cattle using labeled doses of chlortetracycline and oxytetracycline antimicrobials. Vet. Sci. https://doi.org/10.3390/vetsci8110283 (2021).
Jaimes-Duenez, J. et al. Clinical and epidemiological aspects of the infection by Babesia, Theileria and Trypanosoma species in horses from northeastern Colombia. Ticks Tick Borne Dis. 14, 102208. https://doi.org/10.1016/j.ttbdis.2023.102208 (2023).
Koual, R. et al. Phylogenetic evidence for a clade of tick-associated trypanosomes. Parasit. Vectors 16, 3. https://doi.org/10.1186/s13071-022-05622-y (2023).
Homer, M. J., Aguilar-Delfin, I., Telford, S. R. 3rd., Krause, P. J. & Persing, D. H. Babesiosis. Clin. Microbiol. Rev. 13, 451–469. https://doi.org/10.1128/CMR.13.3.451 (2000).
Health., W. O. f. A. Bovine anaplasmosis. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 12th ed. (2023).
Ramos, C. A. et al. Development and assessment of a latex agglutination test based on recombinant MSP5 to detect antibodies against Anaplasma marginale in cattle. Braz. J. Microbiol. 45, 199–204. https://doi.org/10.1590/S1517-83822014005000039 (2014).
Ramos, I. A. S. et al. Serological occurrence for tick-borne agents in beef cattle in the Brazilian Pantanal. Rev. Bras. Parasitol. Vet. 29, e014919. https://doi.org/10.1590/S1984-29612020007 (2020).
Bisen, S. et al. Molecular and serological detection of Anaplasma infection in carrier cattle in north India. Vet. Parasitol. Reg. Stud. Rep. 24, 100550. https://doi.org/10.1016/j.vprsr.2021.100550 (2021).
Carelli, G. et al. Detection and quantification of Anaplasma marginale DNA in blood samples of cattle by real-time PCR. Vet. Microbiol. 124, 107–114. https://doi.org/10.1016/j.vetmic.2007.03.022 (2007).
Li, S. Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, 20. https://doi.org/10.1038/s41421-018-0028-z (2018).
Qiu, M., Zhou, X. M. & Liu, L. Improved strategies for CRISPR-Cas12-based nucleic acids detection. J. Anal. Test. 6, 44–52. https://doi.org/10.1007/s41664-022-00212-4 (2022).
Wang, Y. et al. Ultrasensitive one-pot detection of monkeypox virus with RPA and CRISPR in a sucrose-aided multiphase aqueous system. Microbiol. Spectr. 12, e0226723. https://doi.org/10.1128/spectrum.02267-23 (2024).
Broughton, J. P. et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874. https://doi.org/10.1038/s41587-020-0513-4 (2020).
Wu, J. et al. A CRISPR/Cas12a based universal lateral flow biosensor for the sensitive and specific detection of African Swine-Fever Viruses in Whole Blood. Biosensors (Basel) https://doi.org/10.3390/bios10120203 (2020).
Chen, Q. et al. CRISPR-Cas12-based field-deployable system for rapid detection of synthetic DNA sequence of the monkeypox virus genome. J. Med. Virol. 95, e28385. https://doi.org/10.1002/jmv.28385 (2023).
Ahamed, M. A. et al. Sensitive and specific CRISPR-Cas12a assisted nanopore with RPA for Monkeypox detection. Biosens. Bioelectron. 246, 115866. https://doi.org/10.1016/j.bios.2023.115866 (2024).
Chansiri, L. Tick-borne diseases in Thailand. Trap. Anim. Health Prod. 29, 52S (1997).
Ahantarig, A., Trinachartvanit, W. & Milne, J. R. Tick-borne pathogens and diseases of animals and humans in Thailand. Southeast Asian J. Trop. Med. Public Health 39, 1015–1032 (2008).
Saetiew, N. S. et al. Spatial and seasonal variation in the prevalence of Anaplasma marginale among beef cattle in previously flooded regions of Thailand. Agric. Nat. Resources 54, 355–362. https://doi.org/10.34044/j.anres.2020.54.4.03 (2020).
Arnuphapprasert, A. et al. Genetic characterization of genes encoding the major surface proteins of Anaplasma marginale from cattle isolates in Thailand reveals multiple novel variants. Ticks Tick Borne Dis. 14, 102110. https://doi.org/10.1016/j.ttbdis.2022.102110 (2023).
Junsiri, W. et al. Molecular detection and genetic diversity of Anaplasma marginale based on the major surface protein genes in Thailand. Acta Trop. 205, 105338. https://doi.org/10.1016/j.actatropica.2020.105338 (2020).
Jirapattharasate, C. et al. Molecular detection and genetic diversity of bovine Babesia spp., Theileria orientalis, and Anaplasma marginale in beef cattle in Thailand. Parasitol. Res. 116, 751–762. https://doi.org/10.1007/s00436-016-5345-2 (2017).
Hansmann, Y. et al. Value of PCR, serology, and blood smears for human granulocytic Anaplasmosis diagnosis, France. Emerg. Infect. Dis. 25, 996–998. https://doi.org/10.3201/eid2505.171751 (2019).
Watthanadirek, A. et al. Molecular and recombinant characterization of major surface protein 5 from Anaplasma marginale. Acta Trop. 220, 105933. https://doi.org/10.1016/j.actatropica.2021.105933 (2021).
Watthanadirek, A. et al. Recombinant expression and characterization of major surface protein 4 from Anaplasma marginale. Acta Trop. 197, 105047. https://doi.org/10.1016/j.actatropica.2019.105047 (2019).
Rymaszewska, A. PCR for detection of tick-borne Anaplasma phagocytophilum pathogens—a review. Vet. Med. 56, 529–536 (2011).
Jenkins, C. et al. Detection and identification of bacteria in clinical samples by 16S rRNA gene sequencing: Comparison of two different approaches in clinical practice. J. Med. Microbiol. 61, 483–488. https://doi.org/10.1099/jmm.0.030387-0 (2012).
Lobato, I. M. & O’Sullivan, C. K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Anal. Chem. 98, 19–35. https://doi.org/10.1016/j.trac.2017.10.015 (2018).
Salazar, A., Ochoa-Corona, F. M., Talley, J. L. & Noden, B. H. Recombinase polymerase amplification (RPA) with lateral flow detection for three Anaplasma species of importance to livestock health. Sci. Rep. 11, 15962. https://doi.org/10.1038/s41598-021-95402-y (2021).
Jiang, L., Ching, P., Chao, C. C., Dumler, J. S. & Ching, W. M. Development of a sensitive and rapid recombinase polymerase amplification assay for detection of Anaplasma phagocytophilum. J. Clin. Microbiol. https://doi.org/10.1128/JCM.01777-19 (2020).
Zhang, Y.-M., Zhang, Y. & Xie, K. Evaluation of CRISPR/Cas12a-based DNA detection for fast pathogen diagnosis and GMO test in rice. Mol. Breed. https://doi.org/10.1007/s11032-019-1092-2 (2020).
Liu, H. et al. RPA-Cas12a-FS: A frontline nucleic acid rapid detection system for food safety based on CRISPR-Cas12a combined with recombinase polymerase amplification. Food Chem. 334, 127608. https://doi.org/10.1016/j.foodchem.2020.127608 (2021).
Palaz, F., Kalkan, A. K., Tozluyurt, A. & Ozsoz, M. CRISPR-based tools: Alternative methods for the diagnosis of COVID-19. Clin. Biochem. 89, 1–13. https://doi.org/10.1016/j.clinbiochem.2020.12.011 (2021).
Swarts, D. C. Making the cut(s): How Cas12a cleaves target and non-target DNA. Biochem. Soc. Trans. 47, 1499–1510. https://doi.org/10.1042/BST20190564 (2019).
Paul, B. & Montoya, G. CRISPR-Cas12a: Functional overview and applications. Biomed. J. 43, 8–17. https://doi.org/10.1016/j.bj.2019.10.005 (2020).
Kaminski, M. M., Abudayyeh, O. O., Gootenberg, J. S., Zhang, F. & Collins, J. J. CRISPR-based diagnostics. Nat. Biomed. Eng. 5, 643–656. https://doi.org/10.1038/s41551-021-00760-7 (2021).
Lee, P. Y., Wong, Y. P., Othman, S. & Chee, H. Y. Room-temperature stable loop-mediated isothermal amplification (LAMP) reagents to detect leptospiral DNA. Asian Biomed. (Res. Rev. News) 15, 183–189. https://doi.org/10.2478/abm-2021-0023 (2021).
Patchsung, M. et al. A multiplexed Cas13-based assay with point-of-care attributes for simultaneous COVID-19 diagnosis and variant surveillance. CRISPR J. 6, 99–115. https://doi.org/10.1089/crispr.2022.0048 (2023).
Hayashida, K., Kajino, K., Hachaambwa, L., Namangala, B. & Sugimoto, C. Direct blood dry LAMP: A rapid, stable, and easy diagnostic tool for Human African Trypanosomiasis. PLoS Negl. Trop. Dis. 9, e0003578. https://doi.org/10.1371/journal.pntd.0003578 (2015).
Junsiri, W. et al. Molecular characterization of Anaplasma marginale based on the msp1a and msp1b genes. Vet. Microbiol. 262, 109236. https://doi.org/10.1016/j.vetmic.2021.109236 (2021).
Saisawang, C. et al. Optimal stabilization for long-term storage of nucleic acid-based CRISPRCas12a assay for SARS-CoV-2 detection. Karbala Int. J. Mod. Sci. 9, 187–196. https://doi.org/10.33640/2405-609X.3288 (2023).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. https://doi.org/10.1038/nmeth.2089 (2012).
Acknowledgements
We would like to thank Mr. Idh Pukkanthorn for illustrative work of Fig. 1.
Funding
This research paper is supported by Specific League Funds from Mahidol University. This research was funded in whole, or in part, by the Wellcome Trust [Grant number 220211]. For the purpose of open access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.S., J.W., P.A. and C.S.; investigation: S.S., P.N., A.W. and C.S.; resources: P.A.; writing—original draft: A.S., J.W. and C.S.; writing—review and editing: SDB and C.S.; funding acquisition: C.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sutipatanasomboon, A., Wongsantichon, J., Sakdee, S. et al. RPA-CRISPR/Cas12a assay for the diagnosis of bovine Anaplasma marginale infection. Sci Rep 14, 7820 (2024). https://doi.org/10.1038/s41598-024-58169-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58169-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
