
Abstract
During the process of animal domestication, both natural and artificial selection cause variation in allele frequencies among populations. Identifying genomic areas of selection in domestic animals may aid in the detection of genomic areas linked to ecological and economic traits. We studied genomic variation in 140 worldwide goat individuals, including 75 Asian, 30 African and 35 European goats. We further carried out comparative population genomics to detect genomic regions under selection for adaptability to harsh conditions in local Asian ecotypes and also milk production traits in European commercial breeds. In addition, we estimated the genetic distances among 140 goat individuals. The results showed that among all studied goat groups, local breeds from West and South Asia emerged as an independent group. Our search for selection signatures in local goats from West and South Asia revealed candidate genes related to adaptation to hot climate (HSPB6, HSF4, VPS13A and NBEA genes) and immune response (IL7, IL5, IL23A and LRFN5) traits. Furthermore, selection signatures in European commercial goats involved several milk production related genes, such as VPS13C, NCAM2, TMPRSS15, CSN3 and ABCG2. The identified candidate genes could be the fundamental genetic resource for enhancement of goat production and environmental-adaptive traits, and as such they should be used in goat breeding programs to select more efficient breeds.
Similar content being viewed by others

Introduction
It is believed that domestic goats have been originated from the wild bezoar in the Fertile Crescent and adjacent area1. Their domestication process started about ten thousand years ago in the Neolithic period, just after people's style of living shifted from hunting to farming2. Since then, domestic goat provided hair, fur, meat and milk for human consumption3. Following human migration and trade activities over the past thousand years, domestic goats have been adapted to the environmental conditions under which they have been reared4,5. Today, they comprise over 300 breeds and more than 1006 million individuals, covering indigenous and commercial breeds (http://faostat3.fao.org/browse/Q/QA/E). Throughout the globe, domesticated goats have been adapted to different climate conditions6. For example, in arid regions of Morocco, native goat breeds have acquired traits related to heat tolerance7. In high altitudes of Tibetan Plateau native goats have adapted to the local conditions (such as low-oxygen)8. Moreover, Ugandan native goats have raised their immune merit as a means to abide contamination via parasites in Africa’s hot condition9. These acclimated livestock have prepared a main base for different breeding strategies intended improving selective targets10. For instance, Chinese native goats in the Shandong area have crossed with Saanen dairy goats11 to create Laoshan dairy goats. Due to this attempt, the Laoshan dairy goats have improved to produce high dairy profits and also acquired adaptation to wet weather condition in local area12. In addition, Indonesian native goats in the tropical weather have crossed with Indian native goats11 to create Peranakan Etawah goats. Previous studies have shown that this breed has higher production capacity for producing milk and meat and also adapted to tropical weather conditions13. These studies collectively show that local breeds can survive in harsh conditions and have developed immunity to diseases prevalent in the local conditions.
Over the last few decades, universal climate data perspicuously shows a warming tendency in nearly all areas of the globe, consequent to an extensive dimension of weather changes14. Recently, a considerable diversity of environmental adaptation traits has been detected in different species such as sheep15, goats15,16, chickens17,18 and cattle19,20,21. The current global goat population is 1.002 billion, chiefly existing in Asia (57.7%) and Africa (35.7%), as reported by the Food and Agriculture Organization (FAO), comprising 93.4% of the total number in the world, which has doubled in the last thirty years22. The outstanding countries in goat production included five Asian (China, India, Pakistan, Bangladesh and Iran) and five African (Nigeria, Sudan-former, Sudan, Kenya and Ethiopia) countries22.
Genetic variation in livestock has been acknowledged as a main feature. The preservation of genetic variation is essential in livestock for increasing yield and for answering future issues, which consist of changing environments and food safety23. Long-period natural selection in native goats has led to alterations in the allele frequency and so desirable adaptability as an effect of evolvement, however, commercial goat breeds have been created via a series of extensive artificial selection for increasing production traits. As well, selection signatures identified in domestic animals increase awareness about population demography and will disclose the basis of phenotypic diversity among livestock breeds24.
There are multiple approaches to identifying the footprints of selection. The majority of the accessible approaches use (i) the genetic difference among groups, calculated by FST (fixation index) or related statistics, (ii) the reduce in genetic variation beside footprints of selection in a population25. In this study, whole-genome sequencing (WGS) data from 140 goat individuals, including indigenous ecotypes from Asia (n = 75) and Africa (n = 30), as well as, commercial breeds from European (n = 35) were utilized to characterize the population structure, genetic diversity and signatures of selection analysis. Additionally, we employed nucleotide diversity (θπ) and FST statistical methods to compare Asian local goat individuals (located in West and South regions), as a population that does not undergone artificial breeding programs as yet, with European commercial goat individuals (namely Saanen, Toggenburg and Alpine breeds), to identify potential candidate genes involved in adaptation to harsh environments and milk traits. The candidate genes identified in this work may give a basis for future genome-wide association studies and research into genomic purposes of selection, especially in small ruminants.
Results
Aligning and SNP calling results
The average sequence coverage was 13.19 per sample covering from 5.00442X‐ to 32.46X. In addition, the total number of autosomal SNPs per individual ranged from 5,631,741 to 7,831,222 (Supplementary Table S1).
Population structure, linkage disequilibrium decay and genetic diversity
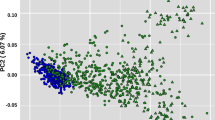
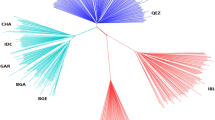
To estimate the phylogeny relationships between studied individuals, a maximum-likelihood (ML) tree was generated. On the basis of this phylogenetic tree (Fig. 1B), the Asian native goat group was separated from other African and European populations. Focusing on the Asian group, samples from Iran, Pakistan and Bangladesh were clustered close to each other. Our findings from Admixture and principal component analysis (PCA) confirm the results of the phylogeny tree (Fig. 1C,D). The PC1 and PC2 accounted for 6.56% and 3.86% of the total genomic variance, respectively. The grouping at = K2 to K5 in admixture output showed the ancestor- ingredient for total individuals investigated (Fig. 1D). The K = 2 separated both African and European populations from the Asian goat population. At K = 3, with the lowest CV error, divides all individuals into three groups, including Asia, Africa and Europe. Ancestral proportions at K = 4 and K = 5 separated the Chinese goat individuals from the other Asian samples. In K = 3–5, Iranian goat samples showed some mixtures with African goat samples (Fig. 1D). In addition, we estimated the diversity (θπ) in each goat population and realized that the Iranian group has higher diversity than other goat groups (Fig. 2A). The amount of linkage disequilibrium (LD) decay between adjoining SNPs throughout the whole genome was calculated to understand the current and classical population size (Ne). The amounts of LD were presented in Fig. 2B (up to 120 kb). The r2 values were the highest in all considered goat groups at marker pair intervals of 1 kb [covered from ~ 0.57 (Iran) to 0.6 0 (Pakistan)] with a slow decrease along with increasing physical intervals between SNPs (up to 20 kb) and again a stable style (> 20 kb). The lowest and highest r2 amounts were observed in Pakistan (0.58) and Africa (0.53) goat groups at marker pairs distance of 120 kb, respectively. Furthermore, we observed that from marker pairs distance of 1 kb to ≤ 20 kb, the decrease in LD was faster in Pakistani goats than other goat groups (Fig. 2B). Concentrating on the Asian goat populations, high r2 values through all genomic distances were observed in Pakistani and Bangladeshi goat ecotypes that are genetically not mixed with the other Asian goat groups (Fig. 1D), whereas the lower r2 amounts were observed in the Iranian goat population (up to 5 kb in physical interval between SNPs), that appear genetically mixed with Africa goat group (Fig. 1D).

(A) geographical areas of studied goat groups. (B) a Phylogenetic tree shows evolutionary relation among different goat breeds (The figure was drawn using Samtools (version 1.31)77, fastree 2 tools (http://www.microbesonline.org/fasttree/) and iTOL program (https://itol.embl.de/)). C. Principal component (PC) analysis (The figure was drawn using GCTA tool (version 1.26.0)80 and R software environment (https://www.r-project.org/)). D. admixture analysis by assuming the ancestral numbers from K = 2 to 5 (The figure was drawn using PLINK program (version 1.9)79, ADMIXTURE program (version 1.3.0)81and R software environment (https://www.r-project.org/)).

Box graphs of nucleotide diversity are calculated for different goat populations (The figure was drawn using VCFtools commands (version 0.1.17)85 and R software environment (https://www.r-project.org/)). The curves of Linkage disequilibrium (LD) in different goat populations (The figure was drawn using Poplddecay program (version 3.42)82 and R software environment (https://www.r-project.org/)).
Genome-wide scan for selection signatures
In this study, we applied whole genome sequence data to perform comparative genome analysis between West and South Asian goat populations as one group and European goat breeds (namely Saanen, Toggenburg and Alpine) as another population to detect selection signatures that are related to various traits. We used nucleotide diversity (Pi) and FST statistics to extract selection signatures remained via natural selection in local goat ecotypes or via artificial selection in commercial European goat breeds. The genomic regions that show extremely high FST values (top 1% for FST) and smaller levels of nucleotide diversity (top 1% for pi) were considered to be selection signatures. Several genes that include significant FST (Fig. 3 and Supplementary Table S2) and nucleotide diversity values (Supplementary Tables S3–S4) were detected in different comparisons. The top FST outlier window (55.00–55.05 Mb) includes genomic signals associated with the VPS13C gene on chromosome 10 (54.90–55.08 Mb), which is related with milk production traits26. Evidence for large negative π scores and high positive FST signals at this genomic region (Fig. 4A) suggest strong positive selection at this locus. Haplotype patterns in this locus throughout all 140 goats are displayed in Fig. 4B. Haplotype pattern in this locus was completely different between commercial goat breeds (European goats) and local goat ecotypes (Asian and African goat ecotypes) (Fig. 4B).

Genomic Manhattan plot of population differentiation by FST, West and South Asian indigenous goat breeds versus European goat breeds, candidate genes linked to adaptation and milk traits are shown with blue and black colors (The figure was drawn using VCFtools commands (version 0.1.17)85 and R software environment (https://www.r-project.org/)).

A. putative sweep area (chr. 10, 55.02–55.04 Mb) is approved by \(\pi\) test (The figure was drawn using VCFtools commands (version 0.1.17)85 and R software environment (https://www.r-project.org/)). B. The patterns of haplotype distribution for VPS13C loci in all 140 goats. The existence of homozygosity and heterozygosity is colored in brown and intermediate brown, respectively. The absence of the derived allele is shown in white. Missing- genotyped regions or individuals are shown in gray (The figure was drawn using Beagle (version 4.0)87, R software environment (https://www.r-project.org/) and python scripts (our in-home script was used)).
Discussion
To estimate genetic affinity among goat groups, we used different analysis methods (Figs. 1 and 2). The genetic divergence and PCA analysis on the basis of the genome data revealed that all Asian goat ecotypes are genetically distinct from the other studied groups. Also, the West and South Asian goat groups were distinguished as an independent population in admixture analysis (k = 3–5). Our results together indicated that the West and South Asian goat populations probably undergo distinct evolutionary processes on the basis of different geographical habituation following domestication and genetic drift, which is consistent with the previous study27. Furthermore, we identified several candidate genes that have been previously reported to be associated with yield phenotypes in goats and other domestic animals. The important genes attributed to adaptation to desert regions, dry weather, and milk traits are shown in Tables 1 and 2. Here we described the relationships between a number of discovered genes and adaptation and milk production traits.
Genes linked to desert regions, dry weather, and adaptation
Indigenous livestock animals have been genetically adapted over generations to their local environmental conditions and are desirable models to investigate the genomic processes underlying adaptability to disease and regional climates. To study this, all West and South Asian indigenous goats were combined to one group and compared to European goats. Previous studies on domestic animals have shown that the heat stress has a negative impact on the production and health, and genomic selection signatures for heat tolerance have recently become an issue for livestock species28. Many studies have been carried out in order to identify genomic variation associated with heat stress in goat breeds15,16. So, the detection of genes linked to heat tolerance can be an answer to issues connected to hot stress in the future. We identified a number of genes engaged in immune response and heat tolerance features through comparative genomic analysis between West and South Asian indigenous goats and European breeds using two high-confidence techniques (greatest 1% FST and 1% log2 θπ ratio amounts) (Table 1). We identified genes related with heat tolerance such as; HSPB6, HSF4, VPS13A and NBEA in West and South Asian indigenous goats (Table 1 and Fig. 3). HSPB6, an example of the most conspicuous component of the HSP group, exists on goat chromosome 18. Kumar et al.29 reported an association between the HSPB6 gene and heat tolerance traits in Indian Karan Fries cattle. Heat shock protein factor 4 was annotated on goat chromosome 1. Xie et al.30 reported that different isoforms of this gene act as activators or inhibitors of tissue specific heat shock gene expression. The VPS13A (Vacuolar Protein Sorting 13 Homolog A) gene encodes chorein, considered a chief moderator of the secretion and density of blood platelets31. Platelet numbers alter total blood density in human beings. Heat stress raises platelet numbers and blood density, which consequently raises the danger of cerebral and coronary thrombosis32. This supports the assumption that the VPS13A gene may play a part in decreasing the danger of thrombosis through regulating platelet numbers and blood density in hot conditions. The VPS13A gene has been discovered in a comparative genomics study of two distinct cattle breeds from Northern and Southern China33. Furthermore, Ai et al.34 discovered that the VPS13A gene plays a role in southern Chinese pig adaptation to hot environments. So far, various homologues of the VPS13 gene associated with adaptation to environmental conditions have been identified in sheep and goats. For example, VPS13B has been reported under selection in the Mediterranean15 and Chinese sheep35 and Moroccan36 and Mediterranean15 goats. Also, the VPS13 C and VPS13D genes have been reported in tropical chicken17 and Mediterranean sheep15. Furthermore, mammalian VPS13 proteins are engaged in caring for lipids37. The VPS13B protein has a role in the formation and development of adipocytes38. In the Maasai, the VPS13D plays a role in cholesterol regulation and lactase persistence39. The Neurobeachin (NBEA) gene, which codes for the neurobeachin protein, was included in selected signatures on chromosome 12. Recently, this gene has been reported in different studies related to heat stress such as Asadollahpour Nanaei et al.18 in Iranian native chickens, Howard et al.40 in beef cattle and Serranito et al.15 in Mediterranean sheep and goats. Furthermore, we identified a number of genes that are likely involved in the goat immune process. Instantly, we detected a class of interleukin (IL7, IL5, IL23A) genes that are related to the immune process. Interleukins are expressed by leukocytes41. Many studies have been conducted to investigate the role of interleukins in the immune system of animals42,43,44. Another gene related to the immune system, the LRFN5 (Leucine Rich Repeat and Fibronectin Type III Domain Containing 5) gene, is located on chromosome 21 of goats, and was found by both FST and Pi methods (top 1%). This gene is involved in immune system response, also known as B-cell mediated immunity45. Previous studies have reported that the LRFN5 gene is associated with adaptation through an immune response in the South African Nguni cattle45 and in indigenous Iranian sheep breeds46. This result is according to preceding researches that reported selection signatures covering immune system genes in indigenous sheep and goats9,47.
Candidate genes associated with milk production traits
The quantity of milk, proteins, and milk fat are particularly important traits in dairy livestock. However, little is known about the area of the genome that controls these important traits in goats. We detected positive selection signatures for milk traits through comparing the genomes of Asian (West and South) indigenous goats with those from European. Whole-genome re-sequencing data from indicative European breeds (namely Saanen, Toggenburg and Alpine) and local native breeds from West and South Asian goats provided a complete list of genomic diversity. Due that European breeds are considered as among the greatest milk yielding in the world54, comparative genome analysis of Asian local breeds and European goats is a desirable approach to detect genomic diversity in milk yield phenotypes. To achieve this purpose, we applied two statistical techniques, including comparisons between two groups and within a group. Protein-encoding genes identified by both FST and nucleotide diversity were reported in Supplementary Tables S2 and S4. We further found a number of genes linked to milk production traits in goats and other ruminants (Table 2). The most notable of them is the VPS13C (Vacuolar Protein Sorting 13 Homolog C) gene that covered the top FST outlier window (on chromosome 10; 55.02–55.04 Mb). Also, low π scores proposed potent positive selection at this location in European goats (Fig. 4A). A number of previous studies have reported the association of the VPS13C gene with milk production traits in goats26 and cattle55. A previous study stated that the VPS13C gene motivates glucose homeostasis for high milk production in cattle55. Furthermore, the different haplotype patterns of the VPS13C gene (chromosome 10; 55,075–55,125 kb) in local goat populations, including Asia and Africa vs European goat population (namely Saanen, Toggenburg and Alpine) suggest the VPS13C gene as a candidate gene related to milk trait in commercial dairy goats. Neural Cell Adhesion Molecule 2 (NCAM2) and Transmembrane Serine Protease 15 (TMPRSS15) genes were identified as candidate genes in a selective sweep region belonging to goat chromosome 1 (top 1% cutoff of FST and Pi methods). Previous works have reported the associations of the NCAM2 and TMPRSS15 genes with fat, protein, and milk yield56,57. Another milk-related candidate gene, Kappa-casein CSN3, was identified in a region located on goat chromosome 1 (top 1% cutoff of FST). The CSN3 gene is an important candidate gene that impacts milk yield traits. Catota-Gómez et al.58 have reported genomic mutations of CSN3 are extremely related to the milk protein percentage. Many independent researchers have reported that its polymorphisms are related to milk yield features (fat, protein and milk production)59 and compound traits (protein and fat percentages)59,60 in different dairy cattle breeds. The ATP Binding Cassette Subfamily G Member 2 (ABCG2) gene was detected as a candidate gene in a selective sweep region belonging to goat chromosome 6 (top 1% cutoff of Pi). A previous study has reported that the ABCG2 gene is strongly related to milk production and composition traits61.
Conclusions
In this study, we discovered several novel and also previously known candidate genes related with milk production traits and adaptability to dry and heat tolerance in goats that can be important for breeding designs. Nevertheless, more research is required to confirm phenotype-genotype connections of the detected genes in this work.
Methods
Genome sequences, short read mapping and SNP calling
Whole genome sequence data of 140 goats from Iran (n = 36), China (n = 30), Pakistan (n = 4), Bangladesh (n = 5), Africa (n = 30) and Europe (n = 35) were downloaded from public sequence databases (https://trace.ncbi.nlm.nih.gov/Traces/sra) (Fig. 1A and Supplementary Table S1).
After quality processing of the raw data, the Burrows–Wheeler Aligner (BWA) program (https://sourceforge.net/projects/bio-bwa)75 was utilized to map the sequence data toward the reference assembly of goat genome (ARS1, GCF_001704415.1)76.
We applied SAMtools commands77 to transform sequence alignment map (SAM) files to binary alignment map (BAM) files. Applying Picard commands (https://github.com/broadinstitute/picard), PCR duplicates were removed from the bam files. Later, in order to raise the quality score for each base, recalibration of the base quality scores was carried out using BaseRecalibrator and IndelRealigner commands from Genome Analysis Toolkit 3.4 (GATK)78. Lastly, SNP discovery and SNP Filtration were done using the UnifiedGenotyper and the Variant Filtration commands the GATK program.
Population structure and genetic ancestry analyses
We used the ML technique to create an evolutionary tree. We applied vcf2fq in vcfutils.pl from Samtools to convert filtered VCF files into FASTA files. The converting of filtered VCF files into FASTA files was done by applying vcf2fq into vcfutils.pl in Samtools and the following utilized FastTree 2 tools to construct a phylogenetic tree. We used the iTOL program (https://itol.embl.de/) for drawing the evolutionary picture. Before the genetic structure analysis, the SNPs data were pruned for LD in PLINK79. Admixture and the PCA were done on pruned SNPs for LD. Genome wide complex trait analysis (GCTA)80 on the basis of SNP genotypes was utilized to specify genetic variation between all goat populations. To study the realizable genomics admixture among groups, we utilized the admixture model applied in the ADMIXTURE program81, applying several values of K (from 2 to 5) and 10,000 iterations. The decay of LD was computed using Poplddecay program82 for different genetic distances between SNP pairs (1, 2.5, 5, 20, 60 and 120 kb).
Statistics to detect selection signatures
Two complementary approaches were applied to explore selection signatures. We computed the genome-wide weighted FST83, because this method is a suitable scale of mean genetic variation between populations having different sizes84. In addition, we calculated diversity (θπ) employing VCFtools commands (‐ window‐pi 50,000 ‐‐window‐pi‐step 25,000)85. Sliding window analyses at the level of the genome were done with a step size of 25 kb and a window size of 50. The log2 (θπ South and West Asia/θπ Europe and θπ Europe/θπ South and West Asia) and mean FST values of SNPs per window were computed. Goat gene IDs that covered all candidate regions were extracted from Ensemble annotation86. To display the specific genotypes patterns of the putative selective region (VPS13C gene on chromosome 10, 55,02–55, 04 Mb), we used Beagle to phase the SNP genotypes and construct haplotype patterns between different populations87. Finally, we showed specific genotypes patterns in a heatmap using python scripts and R software environment.
Data availability
The compiled VCF file is available from the corresponding author upon reasonable request. The other data produced in this work have been presented as supplementary information with this manuscript. The raw sequence data used in this study were downloaded from the public sequence database (https://trace.ncbi.nlm.nih.gov/Traces/sra).
References
Zeder, M. A. & Hesse, B. The initial domestication of goats (Capra hircus) in the Zagros Mountains 10,000 years ago. Science 287(5461), 2254–2257 (2000).
Li, J. & Zhang, Y. Advances in research of the origin and domestication of domestic animals. Biodivers. Sci. 17, 319–329 (2009).
Naderi, S. et al. The goat domestication process inferred from large-scale mitochondrial DNA analysis of wild and domestic individuals. Proc. Natl. Acad. Sci. U. S. A. 105, 17659–17664 (2008).
Taberlet, P. et al. Are cattle, sheep, and goats endangered species?. Mol. Ecol. 17, 275–284 (2008).
Tresset, A. & Vigne, J.-D. Last hunter-gatherers and first farmers of Europe. C. R. Biol. 334, 182–189 (2011).
Nomura, K. et al. Domestication process of the goat revealed by an analysis of the nearly complete mitochondrial protein-encoding genes. PLoS ONE 8, e67775 (2013).
Benjelloun, B. et al. Characterizing neutral genomic diversity and selection signatures in indigenous populations of Moroccan goats (Capra hircus) using WGS data. Front. Genet. 6, 107 (2015).
Song, S. et al. Exome sequencing reveals genetic differentiation due to high-altitude adaptation in the Tibetan cashmere goat (Capra hircus). BMC Genom. 17, 122 (2016).
Onzima, R. B. et al. Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds. Front. Genet. 9, 318 (2018).
Babayan, N. Goat industry development project in ARMENIA. In Sustainable Goat Breeding and Goat Farming in the Central and Eastern European Countries (ed. Sándor, K) 51–56 (FAO, 2014).
Porter, V., Alderson, L., Hall, S. J. G. & Sponenberg, D. P. Mason’s World Encyclopedia of Livestock Breeds and Breeding. (CABI, 2016).
Li, J. Y., Chen, H., Lan, X. Y., Kong, X. J. & Min, L. J. Genetic diversity of five Chinese goat breeds assessed by microsatellite markers. Czech J. Anim. Sci. 53, 315–319 (2008).
Sodiq, A. Doe productivity of kacang and peranakan etawah goats and factors affecting them in Indonesia. Dissertation/doctoral thesis. University of Kassel (2002).
Berg, P., Moseley, C. & Haerter, J. O. Strong increase in convective precipitation in response to higher temperatures. Nat. Geosci. 6, 181–185 (2013).
Serranito, B. et al. Local adaptations of Mediterranean sheep and goats through an integrative approach. Sci. Rep. 11, 1–17 (2021).
Waineina, R. W., Okeno, T. O., Ilatsia, E. D. & Ngeno, K. Selection signature analyses revealed genes associated with adaptation, production, and reproduction in selected goat breeds in Kenya. Front. Genet. 13 (2022).
Tian, S. et al. Genomic analyses reveal genetic adaptations to tropical climates in chickens. Iscience. 23, 101644 (2020).
Asadollahpour Nanaei, H., Kharrati-Koopaee, H. & Esmailizadeh, A. Genetic diversity and signatures of selection for heat tolerance and immune response in Iranian native chickens. BMC Genom. 23, 1–13 (2022).
Yurchenko, A. et al. Scans for signatures of selection in Russian cattle breed genomes reveal new candidate genes for environmental adaptation and acclimation. Sci. Rep. 8, 1–16 (2018).
Goitom, S., Gicheha, M. G., Njonge, F. K. & Kiplangat, N. Landscape genomics and selection signatures of local adaptation of Eritrean indigenous cattle along environmental gradients. Trop. Anim. Health Prod. 53, 1–8 (2021).
Saravanan, K. et al. Genomic scans for selection signatures revealed candidate genes for adaptation and production traits in a variety of cattle breeds. Genomics 113, 955–963 (2021).
Lohani, M. & Bhandari, D. The importance of goats in the world. PAWJ. 6, 9–21 (2021).
Groeneveld, L. et al. Genetic diversity in farm animals—A review. Anim. Genet. 41, 6–31 (2010).
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M. & Wilson, J. F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 19, 220 (2018).
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M. & Servin, B. J. G. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193, 929–941 (2013).
Salgado Pardo, J. I. et al. Candidate genes and their expressions involved in the regulation of milk and meat production and quality in goats (Capra hircus). Animals 12, 988 (2022).
Kim, J. Y. et al. Discovery of genomic characteristics and selection signatures in Korean indigenous goats through comparison of 10 goat breeds. Front. Genet. 10, 699 (2019).
Passamonti, M. M. et al. The quest for genes involved in adaptation to climate change in ruminant livestock. Animals 11, 2833 (2021).
Kumar, R. et al. Genetic polymorphism in HSPB6 gene and their association with heat tolerance traits in Indian Karan Fries (Bos taurus × Bos indicus) cattle. Anim. Biotechnol. 1–12 (2021).
Xie, J. et al. Differential expression of heat shock transcription factors and heat shock proteins after acute and chronic heat stress in laying chickens (Gallus gallus). PLoS ONE 9, e102204 (2014).
Schmidt, E. M. et al. Chorein sensitivity of cytoskeletal organization and degranulation of platelets. FASEB J. 27, 2799–2806 (2013).
Keatinge, W. R. et al. Increased platelet and red cell counts, blood viscosity, and plasma cholesterol levels during heat stress, and mortality from coronary and cerebral thrombosis. Am. J. Med. 81, 795–800 (1986).
Mei, C. et al. Insights into adaption and growth evolution: A comparative genomics study on two distinct cattle breeds from Northern and Southern China. Mol. Ther. Nucleic Acids. 23, 959–967 (2021).
Ai, H. et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 47, 217–225 (2015).
Yang, J. et al. Whole genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Mol. Biol. Evol. 33, 2576–2592 (2016).
Benjelloun, B. Diversité des génomes et adaptation locale des petits ruminants d’un pays méditerranéen: le Maroc (Doctoral dissertation) (Université Grenoble Alpes, 2015).
Gao, L. et al. Gene expression analyses reveal metabolic specifications in acute O2-sensing chemoreceptor cells. Physiol. J. 595, 6091–6120 (2017).
Seifert, W. et al. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem. 286, 37665–37675 (2011).
Wagh, K. et al. Lactase persistence and lipid pathway selection in the Maasai. PLoS ONE 7, e44751 (2012).
Howard, J. T. et al. Beef cattle body temperature during climatic stress: A genome-wide association study. Int. J. Biometeorol. 58, 1665–1672 (2014).
Maggio, M., Guralnik, J. M., Longo, D. L. & Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. Biol. Sci. 61, 575–584 (2006).
Susta, L. et al. Expression of chicken interleukin-2 by a highly virulent strain of Newcastle disease virus leads to decreased systemic viral load but does not significantly affect mortality in chickens. Virol. J. 12, 1–17 (2015).
Chen, W. T. et al. Structure and function of chicken interleukin-1 beta mutants: Uncoupling of receptor binding and in vivo biological activity. Sci. Rep. 6, 27729 (2016).
Truong, A. D. et al. The novel chicken interleukin 26 protein is overexpressed in T cells and induces proinfammatory cytokines. Vet. Res. 47, 65 (2016).
Wang, M. D., Dzama, K., Hefer, C. A. & Muchadeyi, F. C. Genomic population structure and prevalence of copy number variations in South African Nguni cattle. BMC Genom. 16, 1–16 (2015).
Manzari, Z., Mehrabani-Yeganeh, H., Nejati-Javaremi, A., Moradi, M. H. & Gholizadeh, M. Detecting selection signatures in three Iranian sheep breeds. Anim. Genet. 50, 298–302 (2019).
Kim, E. S. et al. Multiple genomic signatures of selection in goats and sheep indigenous to a hot arid environment. J. Hered. 116, 255–264 (2016).
Edea, Z. & Kim, K. S. A whole genomic scan to detect selection signatures between Berkshire and Korean native pig breeds. J. Anim. Sci. Technol. 56, 1–7 (2014).
Jia, P. et al. Four novel SNPs of MYO1A gene associated with heat-tolerance in Chinese cattle. Animals 9, 964 (2019).
Wang, X. et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 6, 1–10 (2016).
Seabury, C. M. et al. Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle. Proc. Natl. Acad. Sci. U.S.A. 107, 151–156 (2010).
Cheon, H. et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32, 2751–2763 (2013).
Estrada-Reyes, Z. M. et al. Signatures of selection for resistance to Haemonchus contortus in sheep and goats. BMC Genom. 20, 1–14 (2019).
Lôbo, A. M. B. O. et al. Characterization of milk production and composition of four exotic goat breeds in Brazil. Small Rumin. Res. 153, 9–16 (2017).
Lemley, C. O., Butler, S. T., Butler, W. R. & Wilson, M. E. Insulin alters hepatic progesterone catabolic enzymes cytochrome P450 2C and 3A in dairy cows. J. Dairy Sci. 91, 641–645 (2008).
Venturini, G. C. et al. Association between single-nucleotide polymorphisms and milk production traits in buffalo. Genet. Mol. Res. 13, 10256–10268 (2014).
Alshawi, A., Essa, A., Al-Bayatti, S. & Hanotte, O. Genome analysis reveals genetic admixture and signature of selection for productivity and environmental traits in Iraqi cattle. Front. Genet. 10, 609 (2019).
Catota-Gómez, L. et al. Frequency and association of polymorphisms in CSN3 gene with milk yield and composition in Saanen goats. Ecosistemasy recursos agropecuarios. 4, 411–417 (2017).
Ogorevc, J., Kunej, T., Razpet, A. & Dovc, P. Database of cattle candidate genes and genetic markers for milk production and mastitis. Anim. Genet. 40(6), 832–851 (2009).
Asadollahpour Nanaei, H., Qanatqestani, D. & Esmailizadeh, A. Whole-genome resequencing reveals selection signatures associated with milk production traits in African Kenana dairy zebu cattle. Genomics 112, 880–885 (2020).
Cohen-Zinder, M. et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 15, 936–944 (2005).
Nayeri, S. et al. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 17, 1–11 (2016).
Ma, J. et al. Assessing the genetic background and selection signatures of Huaxi cattle using high-density SNP array. Animals 11, 3469 (2021).
Buaban, S., Lengnudum, K., Boonkum, W. & Phakdeedindan, P. Genome-wide association study on milk production and somatic cell score for Thai dairy cattle using weighted single-step approach with random regression test-day model. J. Dairy Sci. 105, 468–494 (2022).
Moioli, B., D’Andrea, M. & Pilla, F. J. S. R. R. Candidate genes affecting sheep and goat milk quality. Small Rumin. Res. 68, 179–192 (2007).
Rezvannejad, E., Asadollahpour Nanaei, H. & Esmailizadeh, A. Detection of candidate genes affecting milk production traits in sheep using whole-genome sequencing analysis. Vet. Med. Sci. 8, 1197–1204 (2022).
Illa, S. K., Mukherjee, S., Nath, S. & Mukherjee, A. Genome-wide scanning for signatures of selection revealed the putative genomic regions and candidate genes controlling milk composition and coat color traits in Sahiwal cattle. Front. Genet. 12 (2021).
Gurgul, A. et al. Diversifying selection signatures among divergently selected subpopulations of Polish Red cattle. J. Appl. Genet. 60, 87–95 (2019).
Rajawat, D. et al. Identification of important genomic footprints using eight different selection signature statistics in domestic cattle breeds. Gene 816, 146165 (2022).
Ye, T. et al. Evolutionary analysis of buffalo sterol regulatory element-binding factor (SREBF) family genes and their affection on milk traits. Anim. Biotechnol. 1–12 (2022).
Suarez-Vega, A., Gutierrez-Gil, B. & Arranz, J. J. Transcriptome expression analysis of candidate milk genes affecting cheeserelated traits in 2 sheep breeds. J. Dairy Sci. 99, 6381–6390 (2016).
Sutera, A. M. et al. Genome-wide association studies for milk production traits in Valle del Belice sheep using repeated measures. Anim. Genet. 50, 311–314 (2019).
Bauman, D. E. & Currie, W. B. Partitioning of nutrients during pregnancy and lactation: A review of mechanisms involving homeostasis and homeorhesis. J. Dairy Sci. 63, 1514–1529 (1980).
Shahbazkia, H. R., Aminlari, M. & Cravador, A. Association of polymorphism of the β (1, 4)-galactosyltransferase-I gene with milk production traits in Holsteins. Mol. Biol. Rep. 39, 6715–6721 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. J. Bioinform. 25, 1754–1760 (2009).
Bickhart, D. M. et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650 (2017).
Li, H. et al. The sequence alignment/map format and SAMtools. J. Bioinform. 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M. & Yang, T. L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. J. Bioinform. 35, 1786–1788 (2019).
Weir, B.S. & Cockerham C.C. Estimating F-statistics for the analysis of population structure. J. Evol. 1358–1370 (1984).
Spolaore, E. & Wacziarg, R. The diffusion of development. Q. J. Econ. 124, 469–529 (2009).
Danecek, P. et al. The variant call format and VCFtools. J. Bioinform. 27, 2156–2158 (2011).
Flicek, P. et al. Ensembl. Nucleic Acids Res. 40, D84-90 (2012).
Browning, B. L., Tian, X., Zhou, Y. & Browning, S. R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890 (2021).
Acknowledgements
This work was conducted as postdoctoral research at Shahid Bahonar University of Kerman, Kerman, Iran. Z.A.G. was supported jointly by Iran National Science Foundation (INSF; Grant number: 99013618) and Shahid Bahonar University of Kerman.
Author information
Authors and Affiliations
Contributions
A.A.M. and A.E. led the study. Z.A.G., and H.A.N. analyzed the data. Z.A.G. drafted the manuscript. H.A.N, Z.A.G., A.E., and A.A.M. revised the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amiri Ghanatsaman, Z., Ayatolahi Mehrgardi, A., Asadollahpour Nanaei, H. et al. Comparative genomic analysis uncovers candidate genes related with milk production and adaptive traits in goat breeds. Sci Rep 13, 8722 (2023). https://doi.org/10.1038/s41598-023-35973-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35973-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
