
Abstract
The Japanese rhinoceros beetle Trypoxylus dichotomus is a giant beetle with distinctive exaggerated horns present on the head and prothoracic regions of the male. T. dichotomus has been used as a research model in various fields such as evolutionary developmental biology, ecology, ethology, biomimetics, and drug discovery. In this study, de novo assembly of 615 Mb, representing 80% of the genome estimated by flow cytometry, was obtained using the 10 × Chromium platform. The scaffold N50 length of the genome assembly was 8.02 Mb, with repetitive elements predicted to comprise 49.5% of the assembly. In total, 23,987 protein-coding genes were predicted in the genome. In addition, de novo assembly of the mitochondrial genome yielded a contig of 20,217 bp. We also analyzed the transcriptome by generating 16 RNA-seq libraries from a variety of tissues of both sexes and developmental stages, which allowed us to identify 13 co-expressed gene modules. We focused on the genes related to horn formation and obtained new insights into the evolution of the gene repertoire and sexual dimorphism as exemplified by the sex-specific splicing pattern of the doublesex gene. This genomic information will be an excellent resource for further functional and evolutionary analyses, including the evolutionary origin and genetic regulation of beetle horns and the molecular mechanisms underlying sexual dimorphism.
Similar content being viewed by others


Introduction
Beetles (Insecta: Coleoptera) are the largest order not only among insects but among all animals, and they are regarded as one of the most successful animal groups1. Beetles exhibit extraordinary morphological, ecological, and behavioral diversity2 and have been used as models to study ecological and evolutionary biology for centuries. For example, when Darwin introduced the term "sexual selection," the beetle horn was featured as a typical example3.
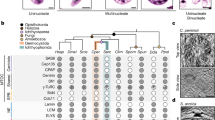
Trypoxylus dichotomus, commonly known as the Japanese rhinoceros beetle, is a giant beetle reaching up to 91.7 mm in length (Fig. 1a). First described by Linnaeus4, T. dichotomus belongs to the Scarabaeidae, Dynastini, Trypoxylus (Fig. 1b). The genus Trypoxylus is most closely related to the genus Xyloscaptes (Fig. 1b)1,5,6,7,8. T. dichotomus inhabits East Asia including Japan, China, Tibet, Taiwan, the Korean peninsula, Northeastern India, Thailand, Vietnam, Laos, and Myanmar. T. dichotomus is characterized by the set of exaggerated horns present in the head and prothoracic regions of males, while females have no horns. The horn of the head region (head horn) can extend to more than 1/3 of the male’s body length and is bifurcated twice at the distal tip. The horn of the prothoracic region (thoracic horn) is shorter and bifurcated once at the distal tip. Both horns are strongly sexually dimorphic, and are used as weapons in combat between males over feeding territories visited by females9. Battles take place on the trunks of host trees, with males inserting their head horn under the prothorax of an opponent and attempting to scoop the rival off of the tree. Biomechanical studies of horn morphology indicate that the shape of the horn, particularly the triangular cross-section of the base of the horn, resists buckling when twisted and is well suited to the nature of the battles in this species10.

Photograph and phylogenetic context of the Japanese rhinoceros beetle, Trypoxylus dichotomus. (a) Photograph of an adult male (left) and an adult female (right) of T. dichotomus. (b) A phylogenetic tree of beetles depicting the phylogenetic relationship between T. dichotomus and related beetles with D. melanogaster as an outgroup. Estimated divergence dates (mya: million years ago) are based on Hunt et al., Ahrens et al., Misof et al., Mckenna et al. and Jin et al.1,5,6,7,8.
T. dichotomus is a univoltine insect. Larvae feed and grow on humus soil or decaying wood from autumn to spring. During the prepupal stage, the larvae form a pupal chamber using a mixture of fecal pellets and humus to pupate below ground. In early summer, the adults appear in broad-leaved forests to ingest sap, and males and females mate on trees near feeding sites. After mating, the female lays the eggs in the humus. Adults have a maximum lifespan of about three months and are unable to overwinter.
T. dichotomus is an emerging model insect with many advantages. First, it is easy to obtain and the breeding/culturing system in the laboratory has been well developed. Larvae are easily maintained in the laboratory and can be stored at low temperatures to delay the onset of pupation, facilitating their use for research throughout the year11. For the critical period from prepupa to adult, when horn formation occurs, a soil-free breeding system has been established that permits non-invasive continuous monitoring and the precise staging of horn development12. Second, an RNA interference (RNAi) technique has been established in T. dichotomus, which allows functional assessment of genes of interest11,12,13,14,15,16,17. Larval RNAi is performed by injecting double-stranded RNA (dsRNA) into the 1st thoracic segment (T1 segment) of the last instar larva just before the prepupal stage. Larval RNAi is so efficient that researchers can carry out large-scale RNAi screens against candidate genes, as proved by our successful identification of genes related to horn formation11,12,13,14,15,16. Third, the large body size of T. dichotomus has advantages in efficient sampling for various experiments, which could facilitate biochemical assays and molecular analyses including next-generation sequencing (NGS)15,18. For example, sufficient amounts of RNA and DNA required for NGS studies can be obtained from each tissue of a single individual alone as shown in this paper.
Studies of T. dichotomus have increased rapidly in a wide variety of research fields due to the utility of this species as a model system. For example, in developmental biology the mechanisms of T. dichotomus horn formation have been described in depth19. The molecular pathways underlying sexual dimorphism of the exaggerated horns have been intensively studied11,12, and numerous genes involved in horn formation have been identified by comprehensive transcriptome analyses15. In addition, beetle horns are considered to be an evolutionary novelty because there is no obvious homologous structure in the ancestral species20, and T. dichotomus horns have proven to be an excellent model for exploring how novel morphological structures arise.
The length of T. dichotomus horns exhibits a highly sensitive response to the nutritional condition of larvae21. Many exaggerated ornaments and weapons of sexual selection exhibit such nutrition- or condition-dependent expression. This plasticity is considered to be an integral component of their function as a reliable signal of the quality of a male. The horns of T. dichotomus were the first sexually-selected structure of any animal species to have these underlying mechanisms of conditional expression explored at a developmental or genetic level13. The resulting variation in male horn length is mildly polyphenic, yielding major and minor males9,22, and ethological studies revealed that males tap each other with their head horns before direct combat23, to assess the size of their competitors and avoid unnecessary fighting24. Less competitive minor males have alternative reproductive strategies to spend more time mating with females25 and appear at feeding sites earlier than major males to avoid fighting and encounter females26,27.
The large size and extraordinary morphology of T. dichotomus has inspired several biomimetic studies. For example, the mechanical properties of elytra (fore wings)28,29,30,31 and the aerodynamic mechanisms32 of T. dichotomus flight have been studied and applied to industrial uses. Furthermore, the high degradation efficiency of T. dichotomus for lignocellulose, which is difficult to degrade, is expected to be effectively utilized in the biotreatment of plant biomass33,34,35,36. In addition, an anti-bacterial peptide37,38,39,40,41,42,43 and a molecule with anti-prion activity44 were discovered from T. dichotomus and medical applications of these molecules are expected. Thus, T. dichotomus has been actively used in various research fields, including evolutionary developmental biology, ecology, ethology, biomimetics, and drug discovery.
Despite the excellent properties of this beetle as a research model, limited genetic information has been available for T. dichotomus until recently, despite a growing number of NGS studies including genomic and transcriptomic analyses being reported15,18,45,46. In the present study, we present the draft genome of T. dichotomus septentrionalis, along with functional annotation and gene expression data. In addition, we compare the gene repertoire of T. dichotomus with those of three other insects. We focus especially on the enlarged male horns of this species, aiming to gain a deeper understanding of the genetic basis of horn formation and the evolution of these genes. This genomic information provides a crucial foundation for future studies using T. dichotomus as well as the comparative genomics of insects.
Materials and methods
Insects
We purchased T. dichotomus septentrionalis larvae from Loiinne (Gunma, Japan). The sex of last (third) instar larvae were determined according to previously described methods11, and the larvae were then individually fed on humus in plastic containers, and kept at 10 °C until use. For tissue sampling, larvae were moved to room temperature for a minimum of 10 days and reared at 28 °C.
Genome sequencing and de novo assembly
Male leg primordia derived from a single individual at 72 h after pupal-chamber formation, which is before cuticle pigmentation and sclerotization, were dissected out from prepupa in 0.75% sodium chloride solution, and used for genome analysis.
To prepare high molecular weight (HMW) DNA of the beetle, 184.5 mg of the frozen tissue was transferred to a mortar and gently ground into a fine powder with liquid nitrogen. Frozen powdery QIAGEN G2 buffer (Qiagen, Netherlands, Cat #1014636), which was generated by spraying the buffer into liquid nitrogen in a glass beaker, was added to the sample and blended quickly. Letting the mixture thaw in a tube, RNase A (QIAGEN, Cat #19101) and Proteinase K (QIAGEN, Cat #1019499) were added, and the sample was incubated at room temperature for 2.5 h without agitation. The sample was centrifuged at 5000 × g for 30 min and the supernatant was subjected to DNA extraction with a QIAGEN Genomic-tip 100/G column (Cat # 10243). The genomic DNA was eluted with 5 ml of Buffer QF, and then purified and concentrated using 0.5-fold volume of Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA, Cat # A63881) yielding 12.2 µg of DNA (174.0 ng/µl). The DNA was size-fractioned by using SAGE HLS (Sage Science, Beverly, MA, USA, Cat #HLS0001), and the fraction with the highest molecular size was collected. The size distribution of the HMW DNA was evaluated by pulsed-field-gel-electrophoresis (PFGE). In brief, 20 ng of HMW DNA was run on a 1% agarose gel (Seakem Gold Agarose, Lonza, Rockland, ME, USA, Cat #50150) in 0.5xTBE with the BioRad CHEF Mapper system (BioRad, Hercules, CA, USA, Cat #M1703650) for 15 h, and the gel was stained with SYBR Gold dye (Thermo Fisher Scientific, Waltham, MA, USA, Cat #11494). Lambda ladder, Saccharomyces cerevisiae genome, and 5 kbp ladder (BioRad, Cat #170–3624) were used as standards. These results demonstrated the HMW DNA had an approximate mean size of 50–80 kbp, while shorter DNA fragments (< ~ 10 kbp) were efficiently removed (Fig. S1). DNA concentration was measured with a Qubit fluorometer (Thermo Fisher Scientific, Cat #Q32866) using the Qubit™ dsDNA BR Assay Kit (Thermo Fisher Scientific, Cat #Q32850).
We constructed a 10 × Genomics Chromium linked-read library using 0.68 ng of the HMW DNA extracted as described above with a Chromium Genome Library Kit & Gel Bead Kit v2 (10 × Genomics, San Francisco, CA, USA, Cat #120258) following the manufacturer's protocol. The generated library was sequenced on a HiSeqX (IIlumina, San Diego, CA, USA) at Macrogen Japan Corp (Tokyo, Japan). The obtained Illumina reads were assembled using the Supernova assembler (ver. 2.1.0)47 with a parameter of maxreads = 265000000.
We assembled the mitochondrial genome of T. dichotomus separately from a paired-end library. Genomic DNA was extracted from the leg primordia using the QIAGEN Genomic-Tip. A paired-end library was prepared with the TruSeq DNA PCR-Free Library Preparation Kit (Illumina) from 1 µg of the genomic DNA. The library was sequenced using the Illumina HiSeqX system with 2 × 150 bp paired-end sequencing protocol at Macrogen Japan Corp (Tokyo, Japan), where 182,102,568 read-pairs were produced. The mitochondrial genome was assembled using a subset of the raw reads (1 M reads, 302 Mb) by NovoPlasty (v4.3.1)48 with a parameter of K-mer = 33, using a partial sequence of 16S rRNA of T. dichotomus (GenBank accession: AB178318.1) as the seed sequence. Our de novo assembly yielded two contigs, Contig1 (20,217 bp) and Contig2 (1163 bp). A manual inspection revealed that Contig2 was a repetitive sequence included in Contig1 and Contig1 corresponds to the mitochondrial genome. Note that the repetitive regions were not fully resolved by the de novo assembly from Illumina short reads and it is likely the contig, in fact, contains more units of the repeats. The assembled contig was annotated using MITOS2 webserver (http://mitos2.bioinf.uni-leipzig.de/index.py)49 with the invertebrate mitochondrial genetic code. The circular map of the mitochondrial genome was drawn using OrganellarGenomeDRAW50.
Genome annotation
Repeat annotation: We constructed the model of repetitive elements in the genome of T. dichotomus, using RepeatModeler (ver. open-1.0.8)51 with the NCBI search engine. The repeat content was identified with RepeatMasker (ver. open-4.0.6)52 using the result of RepeatModeler as a custom library. Other options were not specified.
Gene prediction: We annotated the T. dichotomus genome for protein-coding genes using RNA-seq data. We generated 16 libraries covering 6 tissues and 6 developmental time points from early embryogenesis to postembryonic stages (see RNA-Seq analysis section below). The Illumina RNA-seq reads were aligned against a hard-masked genome in which genomic repetitive elements were substituted by ‘N’, using HISAT2 (ver. 2.1.0)53 with default parameters. The BRAKER2 (ver. 2.1.5)54 pipeline was used to predict protein-coding genes based on the RNA-seq alignments.
Gene annotation: All predicted protein-coding genes were compared with the NCBI non-redundant protein database (nr DB, release November 2020) using the blastp command of diamond software (ver. 2.0.5)55 with a threshold of e-value < 1.0e−5. We used InterProScan (ver. 5.48)56 to query the predicted coding regions for known functional domains and assign Gene Ontology (GO) terms to the proteins. We also used the eggNOG-mapper pipeline (ver. 2.0.1b)57 for functional annotation (including GO term assignments) of the predicted genes based on the orthology information.
BUSCO (ver. 4.0.6)58 was used in quantitative measuring for the assessment of genome assembly, using insecta_odb10 (1367 total orthogroups) as the lineage input. A genome browser was built using JBrowse2 web (ver. 1.5.1)59 and is available at http://www.insect.nibb.info/trydi/.
Ortholog analysis
We used the OrthoFinder (ver. 2.3.11)60 to generate clusters of orthologous and paralogous gene families. Public gene datasets of Onthophagus taurus (RefSeq accession No. GCF_000648695), Tribolium castaneum (UniProt accession No. UP000007266), and Drosophila melanogaster (UniProt accession No. UP000000803) were used as references.
RNA-Seq analysis
In total, 16 samples (egg, non-sexed embryos from five stages, ovary, testis, and Malpighian tubule, hindgut, brain and fat body of males and females, respectively (Table S1)) were used for RNA-seq analysis. Eggs, ovaries, and testes were dissected out from adult insects in 0.75% sodium chloride solution. Malpighian tubule, hindgut, brain and fat body were dissected out from third instar larvae in 0.75% sodium chloride solution. These samples were frozen in liquid nitrogen and stored at − 80 °C until use.
Total RNA was extracted from each tissue sample, except for the fat body sample, using the RNeasy Micro Kit (QIAGEN, Cat # 74004) according to the manufacturer's instructions. Total RNA of the fat bodies was extracted using TRIzol Reagent (Thermo Fisher Scientific, Cat #15596-026) according to the manufacturer's instructions. These total RNAs were treated with DNase using RNase-Free DNase Set (QIAGEN, Cat #79254), from which Illumina sequencing libraries were prepared using the TruSeq Stranded mRNA library prep kit (Illumina) following the manufacturer's instructions. The libraries were multiplexed and sequenced using the Illumina HiSeq1500 system with 2 × 101 bp paired-end sequencing protocol at the Functional Genomics Facility of National Institute for Basic Biology.
T. dichotomus RNA-Seq reads generated from these 16 libraries were adapter-trimmed using Trim Galore! (ver. 0.5.0)61 and cutadapt (ver. 1.18)62. The cleaned RNA-Seq reads were mapped to the T. dichotomus genome assembly using HISAT2 (ver. 2.1.0)53 with default parameters. For quantification of gene expression, StringTie (ver. 2.1.3)63 and prepDE.py (http://ccb.jhu.edu/software/stringtie/dl/prepDE.py) with a GTF file of the BRAKER2 gene prediction were used to generate the count matrix. The count data were normalized by the trimmed mean M values (TMM) method available in the edgeR library (ver. 3.32.1)64,65,66. To visualize profiles of gene expression, a multidimensional scaling (MDS) plot was generated using the edgeR software package.
To detect modules of co-expressed genes from the transcriptome data, Weighted Gene Correlation Network Analysis (WGCNA) was applied67. Normalized count data were used for this analysis implemented in the WGCNA library (version 1.70)68. Genes expressed at low levels, and genes with low expression variance across the libraries, were filtered out; the 6666 surviving gene models were used in the WGCNA analysis. A signed network was constructed in WGCNA with specific parameter settings of power = 8, networkType = "signed", TOMType = "unsigned", minModuleSize = 30.
Genome size estimation using flow cytometry
Flow cytometry estimates were made by quantification of fluorescence from propidium iodide (PI) stained nuclei extracted from primordia of horns, legs or wings of male pupae with the fruitfly, D. melanogaster (1C = 173.3 Mbp) as an internal standard. Samples were added to 1 mL of PBS and homogenized with BioMasher II (nippi, Tokyo, Japan, cat# 320102), and then 1 µl of 10% Triton-X (SIGMA-ALDRICH, St Louis, MO, USA, cat# 93443) and 4 µl of 100 mg/ml RNase A (QIAGEN, cat# 19101) were added to the homogenate. The resultant solution was passed through a 30 µm CellTrics filter (Sysmex Partec, Görlitz, Germany, cat# BP486257) and stained with 10 µg/ml PI (Sony Biotechnology, San Jose, CA, USA, cat# 2706505). The nuclei were analyzed on a Cell Sorter SH800 (SONY Biotechnology) for three replicates.
Genome size estimation from reads of shotgun sequencing
Genomic DNA was extracted from the leg primordia using the QIAGEN Genomic-Tip (Cat # 10243). A paired-end library was prepared with the TruSeq DNA PCR-Free LT Sample Preparation Kit (Illumina, #FC-121-3001) from 1 µg of the genomic DNA. The library was sequenced using the Illumina HiSeqX system with 2 × 150 bp paired-end sequencing protocol at Macrogen Japan Corp (Tokyo, Japan), where 182,102,568 read-pairs were produced. To estimate the genome size of T. dichotomus, we analyzed the distribution of k-mers from the Illumina reads using Jellyfish (ver. 2.2.10)69 and GenomeScope 2.0 (git commit id: fdeb891)70. The distribution of k-mers of size was analyzed with three different k-mer sizes, 21, 31 and 41.
Whole genome alignment
We compared our T. dichotomus assembly with another published assembly derived from the Chinese population, as reported by Wang et al.46. The assembly of T. dichotomus isolate ZJPA, in FASTA format, was downloaded from the publication’s supporting data website on GigaScience (http://gigadb.org/dataset/102226). Whole genome alignment of the two assemblies was conducted using unimap ver. 0.1-r46 (https://github.com/lh3/unimap), a variant of minimap271 optimized for assembly-to-reference alignment, with the following parameters: -x asm10. The generated alignment result, in PAF format, was then analyzed using paftools, a utility program included in the minimap2 distribution, to obtain information on structural variations. Dot plot visualization was conducted with D-Genies72.
Results and discussion
Genome size estimate
Using flow cytometry and the fruitfly D. melanogaster as reference, we determined that the haploid genome size of T. dichotomus is 773.1 ± 24.6 Mb. In addition, we estimated the T. dichotomus genome size from the distribution of k-mers from Illumina reads of shotgun sequences. The distribution of k-mers of size 21, 31 and 41 resulted in an estimated haploid genome size of 710–764 Mb. The small discrepancy between k-mer and cytometry-based estimates may be caused by the repetitive elements (see below), which can affect k-mer estimates.
Genome assembly and evaluation
Genome sequencing of T. dichotomus was performed with genomic DNA isolated from leg primordia of a single male prepupa collected in Gunma, Japan. We prepared high molecular weight (HMW) DNA from the beetle sample with an approximate mean size of 50–80 kbp. The HMW genome DNA (0.68 ng) was subjected to the linked-read whole-genome sequencing library construction on the 10 × Chromium platform. A draft genome was assembled with Supernova47 using 265.02 M Illumina reads (57.0 × coverage). The haploid genome assembly was named TdicSN1.0 and used for downstream analysis.
The final assembly TdicSN1.0 consists of 15,609 scaffolds with an N50 of 8.02 Mb and a total size of 615 Mb (Table 1), covering 80% of the genome. We evaluated the completeness of the assembly by using the benchmarking universal single-copy orthologs (BUSCO)73. The BUSCO analysis showed that our T. dichotomus genome assembly has high coverage of coding regions, capturing 99.4% (98.9% complete, 0.5% fragmented) from the Insecta dataset (version 4.0.6; n = 1367) (Table 2). The score is comparable to the genome of the model beetle T. castaneum74.
Recently, a chromosome-level genome assembly of the Chinese population of T. dichotomus was generated through a combination of ONT long read and Hi-C sequencing, as reported by Wang et al.46. We compared our T. dichotomus assembly of the Japanese population with that of the Chinese population. The dot plot indicated a high degree of congruence in the chromosomal structure (Fig. S2a), with very minor indels and inversions, which confirms the accuracy of both assemblies and also highlights the evolutionary conservation of chromosomal structures between populations. On the other hand, the whole-genome alignment of the genomes of the two populations detected a substantial number of polymorphisms at the single nucleotide resolution. Among the 584,585,878 correspondingly mapped bases, 9,457,239 bases (1.6%) were different (single nucleotide polymorphisms; SNPs) between the two genomes, and 1,308,368 indels were detected (Fig. S2b). Genetic partitioning and divergence have been reported for T. dichotomus according to geographical distribution, with clear morphological differences75. Information on the structural variations identified here will be a useful source for studying the evolution of T. dichotomus through comparative genomics approaches.
We assembled the mitochondrial genome separately from a paired-end library using a known partial sequence of 16S rRNA of T. dichotomus (GenBank accession: AB178318.1) as the seed sequence. Our de novo assembly yielded a contig of 20,217 bp. We identified 2 ribosomal RNAs, 22 tRNAs, and 13 protein-coding genes (Fig. S3). The gene repertoire and the structural arrangement showed typical features of mitochondrial genomes of insects, but the total length of the mitochondrial genome was longer than that of the model coleopteran T. castaneum (15.8 kb (NC_003081.2)) by 4.3 kb. The increased length is mostly due to species specific repetitive elements between the s-rRNA gene and the tRNA-GLN gene (Fig. S3).
Genome annotation
The T. dichotomus genome is rich in repetitive elements, which total 305 Mb and account for 49.5% of the genome assembly (Table S2). Considering the situation that the current assembly covers 80% of the estimated genome size, it is likely that uncovered regions contain more repetitive sequences that are generally difficult to capture by Illumina-based genome assembling. We annotated the T. dichotomus genome for protein-coding genes using RNA-seq data. Aiming for a comprehensive gene identification, we collected RNA samples from a wide variety of tissues of both sexes and developmental stages. We generated 16 libraries derived from 6 tissues (brain, Malpighian tubule, hindgut, fat body, testis and ovary) and from 6 time points of whole embryos from middle to late developmental stages (Table S1). The alignment data of these RNA-seq sequences mapped on the TdicSN1.0 assembly were subjected to the BRAKER2 pipeline to predict protein-coding genes. In total, 23,987 protein-coding genes are predicted in the T. dichotomus genome. Of these, 19,708 (82.6%) encoded proteins exhibiting sequence similarity (e-value < 1.0e−5) to proteins in the NCBI non-redundant protein database. The two most frequent top-hit species corresponded to scarab beetles, Oryctes borbonicus, (6312) and O. taurus (5126), followed by other Coleoptera species such as T. castaneum, Ignelater luminosus, and Nicrophorus vespilloides, which reasonably reflects the phylogenetic position of T. dichotomus (Fig. 1b). We used InterProScan to query the predicted coding regions for known functional domains. We identified 29,549 Pfam motifs76 and 34,594 PANTHER motifs77 in the products of 14,497 and 15,139 T. dichotomus gene models, respectively. We also identified 2389 proteins with secretion potential predicted by the SignalP program78. By integrating protein-domain based and orthology-based approaches using InterProScan and eggNOG-mapper pipelines, 581,644 Gene Ontology terms were assigned to 10,541 genes (43.9%).
Ortholog analysis
To understand the gene repertoire evolution of T. dichotomus, we generated clusters of orthologous and paralogous gene families comparing the T. dichotomus proteome with those of three other insects, O. taurus, T. castaneum and D. melanogaster. O. taurus is a dung beetle belonging to the family Scarabaeidae (Fig. 1b), and represents another model insect for the study of horn polyphenism79. We included T. castaneum as a model Coleoptera and D. melanogaster as a model insect for comparison as outgroups. The OrthoFinder program identified 12,034 orthogroups consisting of 56,656 genes derived from all four insects. The 18,783 T. dichotomus gene models were clustered into 10,291 orthogroups, leaving 5204 genes unassigned to any orthogroups (i.e., orphan genes). Among them, 1350 groups were shared within only three Coleoptera species, while 543 groups were shared within only the horned beetles, O. taurus and T. dichotomus (Fig. 2). These beetle-specific groups may account for the common characteristic traits such as the elytron of the beetle80 and the exaggerated horns of T. dichotomus and O. taurus. We found 967 groups, consisting of 5705 genes, that are unique to T. dichotomus and may account for lineage specific traits (Fig. 2).

Venn diagram of shared and unique orthogroups in four insects. Orthogroups were identified by clustering of orthologous groups using OrthoFinder60.
Transcriptome analysis
We analyzed 16 RNA-seq libraries created from a variety of tissues of both sexes and developmental stages (Table S1). We quantified the expression levels of 23,987 protein-coding genes and profiled the expression patterns across all of the sample tissues. The multidimensional scaling (MDS) plot of the 16 samples depict the transcriptome similarities among the samples (Fig. 3a). Samples derived from the same organs clustered together irrespective of sex differences. Eggs and ovaries also clustered together, as did the transcriptomes of embryos. Notably, embryonic samples were roughly ordered on the plot from earlier to later stages, which may represent the gradual transcriptional progression during embryogenesis of T. dichotomus.

Transcriptome analysis. (a) MDS plot for RNA-Seq gene expression of T. dichotomus tissues, organs and developmental samples. Multi-dimensional scaling (MDS) plot showing relatedness between transcript expression profiles of 16 RNA-Seq libraries. Blue circles represent the expression profiles of male samples, and red circles represent those of female samples, while grey squares represent those of samples whose sex are unknown (i.e., embryos). The labels indicating the tissues and sources are defined as follows: Egg, eggs dissected out from the mature ovary; Tt, testis; Ov, ovary; EmM, middle-stage embryos at the stage of ventral appendage formation; EmL1, late stage embryo at the stage when appendages extend; EmL2, late-stage embryos at the stage when appendages are slightly more developed than EmL1; EmL3, late-embryos at a stage when the tracheal pits can be clearly observed; EmL4, late-embryos at a stage of the full-grown embryo; Mt, Malpighian tubules of third instar larva; Hg, hindgut tube of third instar larva; Fb, fat bodies of third instar larva; Br, brains of third instar larvae. See Table S1 for details of the label description. (b, c) Co-expression gene modules identified using WGCNA are shown. Each circle represents the value of the respective module’s Eigengene. Each module represents genes with highly correlated expression profiles, either in a single organ (b) or in a certain stage of embryogenesis (c). Module names and colors shown above each panel correspond to Fig. S4. The name of the organ or developmental stage in which module genes are preferentially expressed is displayed at the right of the module name in each panel. The number at the top right in each panel indicates the number of genes belonging to the module specified.
We performed Weighted Gene Correlation Network Analysis (WGCNA) to understand the co-expression relationship between genes at a system level. WGCNA identified 13 co-expressed modules from the expression data spanning 16 samples; each module contained 71 to 1205 co-expressed genes (Figs. 3b, c, S4). Each module represents genes with highly correlated expression profiles, either in a single organ (Fig. 3b) or in a certain stage of embryogenesis (Fig. 3c). Out of 13 modules, seven represent organ-specific patterns (Figs. 3b, S4, module 1–7). Both modules 6 and 7 represent fat body preferential expression, but genes of module 6 exhibited male-biased expression while genes in the module 7 exhibited female-biased expression, suggesting sexually differentiated functions of T. dichotomus fat body in the sexes. Six modules represent embryonic expression patterns (Figs. 3c, S4, module 8–13). Genes contained in the module 13 exhibited a constant pattern of expression from middle to late stages.
Horn formation genes
The horns of T. dichotomus provide an excellent model for investigating the evolution of novel morphological structures. Our previous research efforts to identify genes involved in horn formation in T. dichotomus were limited by the lack of genomic information and were confined to genes with well-known functions that were conserved across species11,12,13,14,15,16. However, the availability of a genome resource for T. dichotomus now allows us to take a comprehensive, whole-genome approach to understanding the genetic basis of horn formation, as demonstrated by the following examples.
The analysis of orthologs among four insects (T. dichotomus, O. taurus, T. castaneum and D. melanogaster; see above) revealed that 543 orthogroups were shared between two horned beetles, O. taurus and T. dichotomus, but not with the horn-less beetle T. castaneum. These orthologs may provide insight into the characteristic trait of horns found in both species. While functions of most of these genes are currently unknown, those annotated as transcription factors are of particular interest as they may play a role in regulating the gene regulatory network responsible for horn formation (Table S3).
It is widely acknowledged that gene duplication plays a significant role in the acquisition of evolutionarily novel traits81. In this study, we investigated the potential role of gene duplication in T. dichotomus. Specifically, we focused on 17 genes that have been previously linked to horn formation in this species (abrupt, Bar-H1, CyclinE, dachshund, doublesex, fat, Insulin-like receptor, intersex, Notch, Optix, pannier, Retinal Homeobox, Sex combs reduced, Sox14, Sox21b, Sp8 and Tbx20)11,12,13,14,15,16. We found that all of these genes were single-copy with no evidence of duplication. This suggests that the involvement of these 17 genes in horn formation may have resulted from the co-option of existing genes, rather than gene duplication followed by neo-functionalization.
The availability of genome sequence enables us to gain a deeper understanding of the genomic organization of genes. In T. dichotomus, the sex differentiation gene doublesex (dsx) plays a crucial role in horn formation11, and sex-specific splicing has been identified as a key mechanism for regulating this process12. There are various splicing variants in the T. dichotomus dsx gene (such as dsxFL-1, dsxFL-2, dsxFL-3, dsxFS-1, dsxFS-2, dsxFS-3, dsxM-1, dsxM-2, dsxC-1, dsxC-2 and dsxC-3)11. DsxFLs (female-specific isoforms) and dsxMs (male-specific isoforms) each have roles in repressing and promoting horn formation, respectively. Prior to the availability of the genome sequence, the exon–intron structure of the dsx gene was predicted based on cDNA sequences alone11. However, the alignment of transcripts and the genome reference in the dsx locus has allowed for a more precise determination of the gene structure. We discovered that the dsx locus spans 379 kb on Scaffold 32 and comprises 9 exons (Fig. 4). In addition, we found that the regulation of alternative splicing of dsx genes was more complex than previously inferred from cDNA sequence analysis. For example, the Exon 8 of the dsxM2 gene is 10 bases longer than that of dsxM1, leading to an additional stop codon and a distinct protein being translated. Furthermore, non-sex-specific dsx isoforms (dsxC) are not spliced at Exon 4, a finding that has been observed in other insect species including Apis mellifera, O. taurus, and Gnatocerus cornutus, as well as in T. dichotomus82,83,84. The presence of dsxC in these insects may have been a result of intron retention as in A. mellifera and T. dichotomus. We also found that the only difference between dsxFLs and dsxMs, which are involved in sex-specific horn formation, lies in the splicing pattern of Exon 7. This finding suggests that splicing of Exon 7 is crucial and that this splicing pattern determines the presence or absence of horns.

Genome organization of dsx by alignment with genome sequences. The upper gray box indicates the dsx exons localized at scaffold 32. Information on the T. dichotomus dsx isoforms was obtained from in GenBank sequence data (accession numbers AB744665–AB744675). Black boxes indicate untranslated regions (UTRs), Orange boxes indicate open reading frames (ORFs), and Green and Blue boxes indicate the functional domains, DM (Doublesex/Mab-3 DNA-binding; OD1) and OD2 (Oligomerization domain 2) domains. Exons 1, 2 and 3 were difficult to align due to their short sequence, thus the closest conserved region from Exon 4 was selected. It should be noted that the locus of Exons 1, 2 and 3 is uncertain as a result. The scale bar, representing 200 bp, applies only to the exon region, while the intron regions are arbitrary and not to scale. The exon number is indicated above each exon.
Conclusions
We report the assembly and annotation of the nuclear and mitochondrial genomes of the Japanese rhinoceros beetle T. dichotomus septentrionalis. The high level of completeness and the extensive annotation of this genome, in conjunction with transcriptome data collected from diverse tissues and developmental stages, make this draft assembly an excellent resource for further functional and evolutionary analyses in this emerging model insect. In fact, we used genomic information to determine the precise exon–intron structure of the dsx gene in T. dichotomus, and propose that the splicing pattern of exon 7 is critical in determining the presence or absence of horns. Furthermore, the availability of the genome sequence will expedite the use of functional assays using RNAi and genome editing techniques. These are key tools for experimental investigations of the evolutionary origin and genetic regulation of the exaggerated male horn, and the molecular mechanisms underlying the strong sexual dimorphism in its expression. Other research areas such as ethology, biomimetics and drug discovery will benefit from the continuing advancement of genomic resources. Finally, given that T. dichotomus is one of the most popular insects in Japan, we hope that the genomic resources will facilitate the development of effective population genomic tools to monitor and protect wild populations, which would lead to the establishment of the conservation genomics of the Japanese rhinoceros beetle. These genomic data and a genome browser are available at http://www.insect.nibb.info/trydi/.
Data availability
Data from whole-genome sequencing and transcriptome sequencing have been deposited in the DDBJ database under BioProject accession PRJDB12657. The analyzed data including genome assembly, gene prediction, annotation, and gene expression are available through FigShare (https://doi.org/10.6084/m9.figshare.c.5737754). The genome browser is available at http://www.insect.nibb.info/trydi/.
References
Hunt, T. et al. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science 318, 1913–1916 (2007).
Crowson, R. A. The phylogeny of coleoptera. Annu. Rev. Entomol. 5, 111–134 (1960).
Darwin, C. The Descent of Man, and Selection in Relation to Sex (London, 1871).
Linné, C. Mantissa Plantarum: Generum Editionis VI (Impensis Direct. Laurentii Salvii, 1771).
Ahrens, D., Schwarzer, J. & Vogler, A. P. The evolution of scarab beetles tracks the sequential rise of angiosperms and mammals. Proc. R. Soc. B Biol. Sci. 281, 20141470 (2014).
Misof, B. et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767 (2014).
Mckenna, D. D. et al. The beetle tree of life reveals that Coleoptera survived end-Permian mass extinction to diversify during the Cretaceous terrestrial revolution. Syst. Entomol. 40, 835–880 (2015).
Jin, H., Yonezawa, T., Zhong, Y., Kishino, H. & Hasegawa, M. Cretaceous origin of giant rhinoceros beetles (Dynastini; Coleoptera) and correlation of their evolution with the Pangean breakup. Genes Genet. Syst. 91, 209–215 (2016).
Hongo, Y. Evolution of male dimorphic allometry in a population of the Japanese horned beetle Trypoxylus dichotomus septentrionalis. Behav. Ecol. Sociobiol. 62, 245–253 (2007).
McCullough, E. L., Tobalske, B. W. & Emlen, D. J. Structural adaptations to diverse fighting styles in sexually selected weapons. Proc. Natl. Acad. Sci. 111, 14484–14488 (2014).
Ito, Y. et al. The role of doublesex in the evolution of exaggerated horns in the Japanese rhinoceros beetle. EMBO Rep. 14, 561–567 (2013).
Morita, S. et al. Precise staging of beetle horn formation in Trypoxylus dichotomus reveals the pleiotropic roles of doublesex depending on the spatiotemporal developmental contexts. PLOS Genet. 15, e1008063 (2019).
Emlen, D. J., Warren, I. A., Johns, A., Dworkin, I. & Lavine, L. C. A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science 337, 860–864 (2012).
Gotoh, H. et al. The Fat/Hippo signaling pathway links within-disc morphogen patterning to whole-animal signals during phenotypically plastic growth in insects. Dev. Dyn. 244, 1039–1045 (2015).
Ohde, T. et al. Rhinoceros beetle horn development reveals deep parallels with dung beetles. PLOS Genet. 14, e1007651 (2018).
Adachi, H., Matsuda, K., Niimi, T., Kondo, S. & Gotoh, H. Genetical control of 2D pattern and depth of the primordial furrow that prefigures 3D shape of the rhinoceros beetle horn. Sci. Rep. 10, 18687 (2020).
Sakura, K., Morita, S. & Niimi, T. RNA Interference Method for Gene Function Analysis in the Japanese Rhinoceros Beetle Trypoxylus dichotomus. Bio Protoc. 12, e4396 (2022).
Zinna, R. et al. Sexual dimorphism and heightened conditional expression in a sexually selected weapon in the Asian rhinoceros beetle. Mol. Ecol. 27, 5049–5072 (2018).
Matsuda, K. et al. Complex furrows in a 2D epithelial sheet code the 3D structure of a beetle horn. Sci. Rep. 7, 13939 (2017).
Muller, G. B. & Wagner, G. P. Novelty in evolution: Restructuring the concept.
Emlen, D. J., Lavine, L. C. & Ewen-Campen, B. On the origin and evolutionary diversification of beetle horns. Proc. Natl. Acad. Sci. 104, 8661–8668 (2007).
McCullough, E. L., Ledger, K. J., O’Brien, D. M. & Emlen, D. J. Variation in the allometry of exaggerated rhinoceros beetle horns. Anim. Behav. 109, 133–140 (2015).
Hongo, Y. Appraising behaviour during male-male Interaction in the Japanese horned beetle Trypoxylus dichotomus septentrionalis (Kono). Behaviour 140, 501–517 (2003).
McCullough, E. L. & Zinna, R. A. Sensilla density corresponds to the regions of the horn most frequently used during combat in the giant rhinoceros beetle Trypoxylus dichotomus (Coleoptera: Scarabaeidae: Dynastinae). Ann. Entomol. Soc. Am. 106(4), 518–523 (2013).
Karino, K. & Niiyama, H. Males with short horns spent more time mating in the Japanese horned beetle Allomyrina dichotoma. Acta Ethol. 9, 95–98 (2006).
Siva-Jothy, M. T. Mate securing tactics and the cost of fighting in the Japanese horned beetle, Allomyrina dichotoma L. (Scarabaeidae). J. Ethol. 5, 165–172 (1987).
del Sol, J. F. et al. Population differences in the strength of sexual selection match relative weapon size in the Japanese rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae)†. Evolution 75, 394–413 (2021).
Chen, J., Dai, G., Xu, Y. & Iwamoto, M. Optimal composite structures in the forewings of beetles. Compos. Struct. 81, 432–437 (2007).
Tuo, W., Chen, J., Wu, Z., Xie, J. & Wang, Y. Characteristics of the tensile mechanical properties of fresh and dry forewings of beetles. Mater. Sci. Eng. C 65, 51–58 (2016).
Chen, J. et al. The deformation mode and strengthening mechanism of compression in the beetle elytron plate. Mater. Des. 131, 481–486 (2017).
Chen, J., Zhang, X., Okabe, Y., Xie, J. & Xu, M. Beetle elytron plate and the synergistic mechanism of a trabecular-honeycomb core structure. Sci. China Technol. Sci. 62, 87–93 (2019).
Truong, T. V. et al. Flow visualization of rhinoceros beetle (Trypoxylus dichotomus) in free flight. J. Bionic Eng. 9, 304–314 (2012).
Takeishi, H. et al. Xylanolytic and alkaliphilic Dietzia sp. isolated from larvae of the Japanese horned beetle, Trypoxylus dichotomus. Actinomycetologica 20, 49–54 (2006).
Aizawa, T., Urai, M., Iwabuchi, N., Nakajima, M. & Sunairi, M. Bacillus trypoxylicola sp. Nov., xylanase-producing alkaliphilic bacteria isolated from the guts of Japanese horned beetle larvae (Trypoxylus dichotomus septentrionalis). Int. J. Syst. Evol. Microbiol. 60, 61–66 (2010).
Wada, N. et al. Glycolytic activities in the larval digestive tract of Trypoxylus dichotomus (Coleoptera: Scarabaeidae). Insects 5, 351–363 (2014).
Wada, N. et al. Site-specific profiles of biochemical properties in the larval digestive tract of Japanese rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae). Entomol. Sci. 23, 33–43 (2020).
Miyanoshita, A. et al. Isolation and characterization of a new member of the insect defensin family from a beetle, Allomyrina dichotoma. Biochem. Biophys. Res. Commun. 220, 526–531 (1996).
Saido-Sakanaka, H., Ishibashi, J., Sagisaka, A., Momotani, E. & Yamakawa, M. Synthesis and characterization of bactericidal oligopeptides designed on the basis of an insect anti-bacterial peptide. Biochem. J. 338, 29–33 (1999).
Saido-Sakanaka, H., Ishibashi, J., Momotani, E., Amano, F. & Yamakawa, M. In vitro and in vivo activity of antimicrobial peptides synthesized based on the insect defensin. Peptides 25, 19–27 (2004).
Koyama, Y. et al. Cytotoxicity and antigenicity of antimicrobial synthesized peptides derived from the beetle Allomyrina dichotoma defensin in mice. Int. Immunopharmacol. 6, 1748–1753 (2006).
Iwasaki, T., Ishibashi, J., Kubo, M., Taylor, D. & Yamakawa, M. Multiple functions of short synthetic enantiomeric peptides based on beetle defensins. Biosci. Biotechnol. Biochem. 73, 683–687 (2009).
Iwasaki, T. et al. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on the cell surface. Peptides 30, 660–668 (2009).
Iwasaki, T. et al. In vitro activity of diastereomeric antimicrobial peptides alone and in combination with antibiotics against methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa. J. Insect Biotechnol. Sericology 76, 25–29 (2007).
Hamanaka, T. et al. Anti-prion activity found in beetle grub hemolymph of Trypoxylus dichotomus septentrionalis. Biochem. Biophys. Rep. 3, 32–37 (2015).
Ogata, N. Whole-genome sequence of the Trypoxylus dichotomus Japanese rhinoceros beetle. MicroPublication Biol. https://doi.org/10.17912/micropub.biology.000487 (2021).
Wang, Q., Liu, L., Zhang, S., Wu, H. & Huang, J. A chromosome-level genome assembly and intestinal transcriptome of Trypoxylus dichotomus (Coleoptera: Scarabaeidae) to understand its lignocellulose digestion ability. GigaScience 11, giac059 (2022).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw955 (2016).
Donath, A. et al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552 (2019).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64 (2019).
Smit, A. & Hubley, R. RepeatModeler Open-1.0 (2008).
Smit, A., Hubley, R. & Green, P. RepeatMasker. Open-4.0 (2013).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinform. 3, lqaa108 (2021).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354 (2021).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 227–245 (Springer, 2019).
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J. & Holmes, I. H. JBrowse: A next-generation genome browser. Genome Res. 19, 1630–1638 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Krueger, F. Trim Galore (RRID: SCR_011847). https://github.com/FelixKrueger/TrimGalore (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Sun, J., Nishiyama, T., Shimizu, K. & Kadota, K. TCC: An R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 14, 219 (2013).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, 1–45 (2005).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Cabanettes, F. & Klopp, C. D-GENIES: Dot plot large genomes in an interactive, efficient and simple way. PeerJ 6, e4958 (2018).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Herndon, N. et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics 21, 47 (2020).
Yang, H. et al. Phylogeny and biogeography of the Japanese rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae) based on SNP markers. Ecol. Evol. 11, 153–173 (2021).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Mi, H. et al. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, D394–D403 (2021).
Nielsen, H., Engelbrecht, J., Brunak, S. & von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10, 1–6 (1997).
Moczek, A. P. & Kijimoto, T. Development and evolution of insect polyphenisms: Novel insights through the study of sex determination mechanisms. Curr. Opin. Insect Sci. 1, 52–58 (2014).
Grimaldi, D., Engel, M. S., Engel, M. S. & Engel, S. C. Evolution of the Insects (Cambridge University Press, 2005).
Force, A. et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 (1999).
Cho, S., Huang, Z. Y. & Zhang, J. Sex-specific splicing of the honeybee doublesex gene reveals 300 million years of evolution at the bottom of the insect sex-determination pathway. Genetics 177, 1733–1741 (2007).
Kijimoto, T., Moczek, A. P. & Andrews, J. Diversification of doublesex function underlies morph-, sex-, and species-specific development of beetle horns. Proc. Natl. Acad. Sci. 109, 20526–20531 (2012).
Gotoh, H. et al. Molecular cloning and functional characterization of the sex-determination gene doublesex in the sexually dimorphic broad-horned beetle Gnatocerus cornutus (Coleoptera, Tenebrionidae). Sci. Rep. 6, 29337 (2016).
Acknowledgements
We thank Toshiya Ando, Taro Nakamura and Yasuhiko Chikami for helpful discussion, the Model Organisms Facility (Model Plant) / Data Integration and Analysis Facility / Emerging Model Organisms Facility / Trans-Omics Facility of NIBB Trans-Scale Biology Center for technical assistance. This work was supported by MEXT KAKENHI Grant Numbers 23128505, 25128706, 16H01452, 18H04766, 20H04933 and 20H05944 (to Te. N.); 17H06384 (to S. S.); 22128008 (to S. S. and To. N); JSPS KAKENHI Grant Numbers 19K16181 and 21K15135 (to Shi. M.), and NSF IOS–1456133 (to D. E. and L. L.).
Author information
Authors and Affiliations
Contributions
Shi.M., Te.N. and S.S. conceived the study. Shi.M., K.T. and M.N. collected and prepared the tissue samples for sequencing. T.S., To.N., Y.K., K.Y. and S.S. led the bioinformatics analyses. Shi.M., Y.K., K.Y. and S.S. performed the transcriptome sequencing and analysis. K.Y. and S.S. constructed the database. T.B., T.M., Sho.M. and R.C. collected the estimated genome size. Shi.M., T.S., To.N., Y.K., K.Y., T.O., H.G., T.K., J.W., M.S., M.M., K.S., L.L., D.E. and S.S. contributed to the data curation. Shi.M., Te.N. and S.S. wrote and all authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morita, S., Shibata, T.F., Nishiyama, T. et al. The draft genome sequence of the Japanese rhinoceros beetle Trypoxylus dichotomus septentrionalis towards an understanding of horn formation. Sci Rep 13, 8735 (2023). https://doi.org/10.1038/s41598-023-35246-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-35246-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
