
Abstract
Deep-sea mining of hydrothermal deposits off the coast of Japan is currently under consideration, and environmental baseline studies of the area are required to understand possible impacts. The aim of this study is to clarify population structures of dominant benthic megafaunal species near hydrothermal vent fields in the Okinawa Trough, using a population genetics approach. We examined dominant deep-sea scavenging species including eels, several amphipods, and a decapod and performed population genetic analyses based on the mitochondrial cytochrome c oxidase subunit I region. Several sites were sampled within Okinawa Trough to examine intra-population diversity while two other locations 1400–2400 km away were chosen for inter-population comparisons. For synaphobranchid eels Simenchelys parasitica and Synaphobranchus kaupii, our results showed significant intra-population diversity but no inter-population genetic differentiation, suggesting strong genetic connectivity and/or large population sizes. In addition, single nucleotide polymorphism analysis also confirmed strong genetic connectivity for Simenchelys parasitica. Among scavenging amphipods, we detected seven putative species using molecular phylogenetic analysis. We evaluated population structures of the most abundant species of amphipods and a decapod species (Nematocarcinus lanceopes). Our results provide basic information on the genetic population structures of benthic megafaunal species near hydrothermal vent fields, which can be used to select candidate species for future connectivity analysis with high-resolution genetic markers and aid understanding of the potential population impacts of environmental disturbances.
Similar content being viewed by others

Introduction
Resource developers and those involved in biodiversity conservation have raised numerous concerns about the extraction of seafloor minerals1. One such resource, seafloor massive sulfides (SMS), is associated with deep-sea hydrothermal vents2,3,4. However, the chemosynthetic environments of hydrothermal ecosystems have fostered endemic fauna that are also a potential target for conservation. While vent communities themselves have been studied extensively, the marine fauna near hydrothermal vent fields have received little attention in comparison5. The exploitation of SMS is likely to release suspended particles and heavy metals that will not only affect vent communities but also fauna near hydrothermal vent fields that are not directly associated with them, hereafter referred to as “near-vent organisms”. The potential effects of such a scenario are still not well understood.
To achieve a stable supply of mineral resources for the country, the Japan Oil, Gas and Metals National Corporation (JOGMEC) is exploring the extraction of SMS resources, including environmental impact assessments from mining activities. Previous studies have suggested that deep-sea mining of SMS may result in decreases in biodiversity through removal of habitat, release of toxic metals, and burial of organisms from sedimentation [e.g.,5,6,7], among other impacts. To understand environmental impacts from exploitation of SMS, JOGMEC started a technical survey project in 2008 to explore massive sulfide deposits in a seafloor depression near Okinawa in southwest Japan8. JOGMEC performed multiple biological, physical, and geochemical surveys in accordance with guidance provided by the International Seabed Authority9. This included a baseline survey to collect near-vent organisms from the Okinawa Trough.
Information on genetic diversity and connectivity patterns (i.e., determining genetic population structure) is useful for estimating how populations of near-vent organism communities could recover from the impact of mineral extraction10. Megabenthos are a key component of benthic communities, and there have been a relatively large number of studies using population genetic analyses of megabenthos inhabiting hydrothermal vents [e.g.,11]. Because hydrothermal vent distributions are variable and distances between vents can be far apart, it has been suggested that megabenthos inhabiting hydrothermal vents have considerable dispersal ability12,13,14. On the other hand, studies on population genetics of near-vent organisms are still limited. Therefore, understanding the population structure of near-vent organisms, in combination with studies on vent organisms provides important insights into the formation and maintenance mechanisms of hydrothermal ecosystems and their surrounding environments as well as for assessing the impacts of future resource exploitation.
Given the above context, we performed population genetic analyses of several dominant scavenging near-vent megabenthos in the Okinawa Trough based on the mitochondrial cytochrome c oxidase subunit I (COI) region. The mitochondrial COI region is used for relatively high-resolution analysis of interspecies- and intraspecies-level structure15, and it is often used for comparing genetic population structures among deep-sea species11,16. Such comparisons are informative to infer whether a species is sensitive to environmental disturbances as small population/species ranges are generally associated with higher sensitivity. We also conducted population analyses using larger sets of single nucleotide polymorphisms (SNPs) for a single eel species to provide greater resolution and a more comprehensive understanding of genetic structure. Finally, in conjunction with our findings we discuss possible future research efforts targeting near-vent organisms needed for SMS mining to occur.
Materials and methods
Sampling and DNA extraction
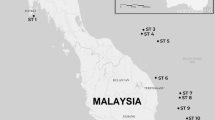
We collected specimens of several deep-sea benthic scavengers including two synaphobranchid eel species, Simenchelys parasitica Gill, 187917 (n = 102) and Synaphobranchus kaupii Johnson, 186218 (n = 6), one decapod species, Nematocarcinus lanceopes Spence Bate, 188819 (n = 23), and multiple amphipod species (n = 43) using baited traps (shrimp pot, conger tube) with Pacific saury during cruises conducted to carry out a technical survey of seven locations in the Okinawa Trough (OT) between 2013 and 2015 (Fig. 1 and Table 1). Decapods were attracted by baited traps and collected with a sledge net. Si. Parasitica, Sy. kaupii, and N. lanceopes were first identified based on morphology. Collected specimens were preserved in a – 20 ºC freezer on board and upon return to land before experiments.

Map showing sampling sites off of Japan. OT = Okinawa Trough, YZ = Suruga Bay, and HD = offshore Hokkaido. This map was made with Natural Earth (free vector and raster map data, https://www.naturalearthdata.com, 1:10 m Cross-blended Hypsometric Tints, version 2.0.0) using the free and open source QGIS version 3.18.1 (https://qgis.org/en/site/).
For the two synaphobranchid eel species, we also examined specimens from two regions outside the Okinawa Trough using organisms from markets to estimate connectivity across large geographic distances; offshore of Hokkaido (HD; n = 25 for Sy. kaupii) and Suruga Bay (YK; n = 6 for Sy. kaupii, n = 24 for Si. parasitica) near Shizuoka Prefecture, Japan (Fig. 1 and Table 1). Specimens of Sy. kaupii from Hokkaido were purchased in a fish market (collected nearby in the Sea of Okhotsk). The distances are ~ 1400 km between the Okinawa Trough and Suruga Bay and ~ 2400 km between the Okinawa Trough and Hokkaido.
DNA extraction was performed using the DNeasy Blood and Tissue kit (QIAGEN, Hilden, Germany) from tissues of preserved specimens according to the manufacturer’s protocol. Extracted DNA was checked by NanoDrop (ThermoFisher Scientific, Waltham, MA, USA) and quantified by Qubit dsDNA HS assay kit (ThermoFisher Scientific, Waltham, MA, USA).
PCR and sequencing
To determine mitochondrial COI sequences, we performed PCR by 20 μL mixture containing 0.5 or 1.0 μL of DNA template, 0.5 μL forward primer (20 μM), 0.5 μL reverse primer (20 μM), 1.6 μL dNTP, 2 μL 10X ExTaq buffer, 0.1 μL ExTaq HS (Takara Bio Inc., Otsu, Japan), and 14.8 or 14.3 μL distilled water. We first used universal primers LCO1490 and HCO2198 and PCR condition: 30 cycles of 0.5 min at 94 °C (denaturation), 1 min at 50 °C (annealing), and 1.5 min at 72 °C (extension), followed by an additional extension for 5 min20. PCR extensions failed for eels, so we designed new primer sets (Table S1) based on a mitogenome sequence of Si. parasitica (accession no. NC_013605). The PCR conditions are as follows: 1 min at 94 °C, 35 cycles of 0.5 min at 94 °C (denaturation), 0.5 min at 60 °C (annealing), and 1 min at 72 °C (extension), followed by an additional extension for 10 min. For amphipods, in addition to universal primers above, we used GrajapCOIF and GrajapCOIR21 and a new primer set (Table S1) designed manually on the basis of a mitochondrial COI sequence of Lysianasoidea sp. (accession no. EF989712). PCR conditions are as follows: 1 min at 94 °C, 35 cycles of 0.5 min at 94 °C (denaturation), 0.5 min at 45 °C (annealing), and 1 min at 72 °C (extension), followed by an additional extension for 10 min.
Each PCR product was cleaned with ExoSAP-IT (Affymetrix, Santa Clara, CA, USA) following the manufacturer’s protocol for direct sequencing. The primers used for the sequencing were the same as those for the PCR amplification (both forward and reverse primers). The purified PCR products were sequenced using the ABI 3730xl DNA Analyzer (Applied Biosystems, CA, USA). Subsequently, partial sequences of COI were obtained through checking the DNA chromatograms by eye and used for the analyses below. Sequence identity was confirmed by NCBI BLASTN. Nucleotide sequences were translated into amino acid sequences to check for the presence of stop codons. Sequence data obtained in this study were deposited in the DNA Data Bank of Japan with the accession nos. LC532844–LC533073.
For Simenchelys parasitica, SNPs were obtained using the protocol of multiplexed ISSR genotyping by sequencing (MIG-seq)22 from 12 to 16 specimens from each site (Table S2). MIG-seq is known as a relatively easy method to evaluate SNPs22. Briefly, we amplified regions of genome DNA around inter-simple sequence repeat (ISSRs) by using universal primer pairs (MIG-seq primer set 1) for the 1st PCR. Then, we pooled DNA libraries with different indexes added by the 2nd PCR and sequenced by DNBSEQ-G400 (MGI Tech.) as paired-end reads (2 × 100 bp). All fastq files have been deposited in the DDBJ database (accession no. DRA014289).
Bioinformatics
For all species examined in this study, we processed sequence data by using custom R code, software version 4.0.323, running packages including ape [functions: read.dna, dist.dna, haploNet;24 and others maintained by the Bioconductor project (https://www.bioconductor.org/), after alignment with MAFFT v7.40225 with the default settings in each species. We also used SeqKit Version: 0.8.126 to obtain basic information (sequence length, the number of sequences) on the FASTA files used in this study. We constructed haplotype networks and calculated genetic diversities by using the R package pegas [functions: haplotype, hap.div, nuc.div;27 under default settings. We also performed analysis of molecular variance (AMOVA) with 1,000 permutations based on R package pegas [function: amova;27 using two (Okinawa Trough and Suruga Bay) populations of Si. parasitica (specimens of OT1–OT7 collected in 2014 and 2015 were pooled as one population because these were all within ~ 5 km of one another; Table 1) and three populations (Okinawa Trough, Suruga Bay, and offshore of Hokkaido) of Sy. kaupii (again all samples in the Okinawa Trough were pooled as a single population; Table 1).
For deep-sea amphipods, Neighbor-Joining (NJ) trees were constructed on genetic distances of Kimura 2-parameter models by using MEGA 728 with 1,000 bootstrap replicates. A model test was performed using ModelTest-NG version 0.1.629, and maximum likelihood (ML) analysis was performed using RAxML-NG version 1.0.330 with 1,000 bootstrap replicates under a GTR (General Time Reversible) + G + I model. Bayesian inference (BI) of phylogenetic analysis was performed using MrBayes v3.2.731 under a GTR + G + I model and 1 MCMC chain (1,000,000 generations and 300,000 for burn-in at which the average standard deviation of split frequency was steadily below 0.01). To identify molecular operational taxonomic units (MOTUs) of deep-sea amphipods, we used Assemble Species by Automatic Partitioning (ASAP)32 based on K80 distance model.
For Si. parasitica, we filtered reads obtained by MIG-seq with FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html) using a fastq-quality-filter v0.0.13 (–Q 33 –q 30 –p 40). Adapter sequences were removed by Cutadapt v2.533. Then we removed shorter reads (< 40 bp) using Seqkit v0.9.126. SNPs were called using the denovo_map.pl pipeline implemented in Stacks v2.034. The parameters used in stacks were as follows: ustacks (-m 3 -M 4) and cstacks (-n 4). Then, we made genepop files using populations (–min-maf 0.03 –max-obs-het 0.5, and -r 0.70) of Stacks. Biallelic loci were filtered with Plink version 1.935 and loci showing deviation from Hardy–Weinberg equilibrium (p < 0.001) and minor allele frequencies (–maf 0.03) were filtered. We obtained basic parameters of genepop files using the package “adegenet” v2.1.336 in R v4.0.323. Principal component analysis (PCA) was also performed with a matrix of individual genotype frequencies using R package “hierfstat”37. In addition, Permutational Multivariate Analysis of Variance (PERMANOVA) with 1st–4th PCs based on Euclidean distances and 999 permutations was performed using the adonis function in the R package vegan38.
Results
We obtained a 767-bp COI sequence of Simenchelys parasitica (BLASTN top hit to nt database: the same species, accession no. AP010849, identity: 99%); a 591-bp sequence from Synaphobranchus kaupii (BLASTN top hit to nt database: the same species, accession no. JF952873, identity: 99%); and a 669-bp sequence from Nematocarcinus lanceopes (BLASTP top hit to nr database: the same species, accession no. ABQ43464, identity: 99%).
Our data indicated high genetic diversity in Si. parasitica populations (Okinawa Trough and Suruga Bay; haplotype diversity = 0.92; Table 2) as well as within population (haplotype diversity > 0.85; Table 3). The AMOVA results indicated that there was no significant genetic differentiation among populations of Si. parasitica (p = 0.4675; Table S3). We also obtained genetic information on three populations of Sy. kaupii (Okinawa Trough, Suruga Bay and Hokkaido; Tables 2, 4) and found relatively low genetic diversity in Hokkaido (haplotype diversity = 0.69; Table 4), while high genetic diversity was found within the other two sites (haplotype diversity > 0.93; Table 4). The AMOVA results showed no significant genetic differentiation among Sy. kaupii populations (p = 0.4805; Table S3). Haplotype networks showed a star-like topology indicating rapid range expansion in both synaphobranchid eel species (Fig. 2). In addition, the MIG-seq analysis detected 110 SNP loci that are available for population genetic analyses for Si. parasitica after filtering random biallelic loci with Plink. PCA indicated no grouping among different populations (Fig. 3) based on individual genotype frequencies. Furthermore, PERMANOVA did not find any significant differences of PCAs among five sites (pseudo F = 1.299, p = 0.201).

Haplotype networks of synaphobranchid eels. (a) Simenchelys parasitica. (b) Synaphobranchus kaupii. Each color corresponds to a population source.

Plot of 1st and 2nd axes of principal component analysis based on a matrix of individual genotype frequencies of Simenchelys parasitica. Abbreviations; Okinawa Trough (OT), Suruga Bay (YZ).
The 23 sequences of N. lanceopes included 11 haplotypes (Table 2; BLASTP top hit to nr database: N. lanceopes, accession no. ABQ43464, identity: 99%). The level of genetic diversity was relatively moderate (haplotype diversity = 0.80, Table 2), and the haplotype network also showed a star-like topology (Fig. 4b).

Haplotype networks of sampled benthos, including (a) Amphipoda sp. (Clade I of Fig. 5), (b) Nematocarcinus lanceopes. Each color corresponds to an individual source.
The amphipod specimens seemed to include several species. Therefore, after extracting 19 haplotypes from 43 sequences (570–573-bp; Table 2, S4), we performed BLASTN with all haplotypes to nt database. The results indicated that 13 haplotypes belonged to the superfamily Lysianassoidea Dana, 184939 (Table S5). We used the similar sequences of each haplotype to perform a molecular phylogenetic analysis, the results of which suggested the existence of seven putative species, as supported by high bootstrap probabilities (NJ = > 99%, ML = > 94%, BI = > 99%; except for Clades II and IV; Fig. 5) and ASAP (asap-score = 1.50). We grouped 25 sequences that putatively belonged to a single species resembling Schisturella pulchra (Hansen, 1888)40 (Clade I, Fig. 5; Table S5) into one population in the Okinawa Trough, showing relatively low genetic diversity (haplotype diversity = 0.43; Table 2). We also constructed a haplotype network for Clade I; the network had a typical star-like topology (Fig. 4a).

Molecular phylogenetic tree of amphipod species. Numbers indicate bootstrap values of neighbor-joining and maximum likelihood methods (only those > 70% are shown) and Bayesian posterior probabilities (shown as NJ/ML/BI). Boxes show clades containing the haplotypes we obtained. Scale indicates 0.05 substitutions per site.
Discussion
Much attention has been given to active deep-sea hydrothermal vents and the unique communities present in these habitats13,41,42, but it is important to evaluate connectivity not only for vent organisms but also for the common deep-sea biota that inhabit the hydrothermal vicinity or “near-vent” organisms. In this study, we described the genetic population structures of several dominant megabenthic organisms (two synaphobranchid eels, one decapod shrimp, and multiple scavenging amphipods) near-vent fields attracted by baited traps. While the species attracted by baited traps may make up a small percentage of the diversity in an area, they can represent a large majority of the abundance as measured with trawl catches43. Synaphobranchid eels are dominant components of the scavenging community in the deep sea44,45,46. We performed genetic analyses on populations of far-flung synaphobranchid eel populations > 2000 km apart, but we could not detect significant differentiation in either eel species among the various sites. This indicates that both species may disperse over large distances (over 1000 km through several generations) and maintain large populations. Some Synaphobranchus species possess polycyclic ovaries47, suggesting frequent opportunities for sexual reproduction among their congeners, which would increase the opportunity for random mating by many individuals. In addition, it is reported that Synaphobranchus kaupii shows high swimming and metabolic activity48, as this species has even been found in abundance in different ocean basins49. Thus, these strategies on life history may serve to increase genetic exchange among populations.
Deep-sea shrimps are also often attracted by baited cameras, and we collected and analyzed 23 individuals of the species Nematocarcinus lanceopes. The haplotype network of N. lanceopes showed a typical star-like topology (Fig. 4b), but the topology was more complicated than those of the other species networks we examined. Such interspecific differences of population structure are also reported from hydrothermal vent shrimp species in the same area11. Future studies should evaluate more individuals and populations to infer whether these topological patterns apply to this species. Population genetic analysis using microsatellite markers has also been attempted in N. lanceopes50,51,52, but the target populations in previous studies were obtained around Antarctica. It is necessary to verify beforehand whether the target population is the same as our target species and whether the same microsatellite markers can be applied.
Roughly 80% of the amphipods examined in this study belonged to the superfamily Lysianassoidea, which is ubiquitous in the deep sea53. Haplotypes VIII, X, and XI in Clade V (Fig. 5) were similar to Amphipoda sp. (accession no. KX365239), but the neighboring haplotypes (accession nos. KX365238, KP713889, and KP713890) were all Abyssorchomene. Thus, these haplotypes likely belong to the genus Abyssorchomene. Our samples contained relatively large numbers of individuals in Clades I and VI (25 and 7 individuals, respectively). Therefore, these two amphipod clades can be considered as good targets for future connectivity analysis focused on organisms near hydrothermal vent fields, at least those in Okinawa Trough. The availability of a relatively large number of amphipod samples makes them suitable for conducting reliable population genetic analyses. Our population analysis of individuals in Clade I of amphipod (Fig. 5), which was similar to Schisturella pulchra (Table S5), showed relatively low genetic diversity among our samples (Table 2). It should be noted that this is likely partly the result of examining only one population. Thus, further studies including additional sites are needed to evaluate the geographic patterns in genetic diversity in this amphipod clade. It should also be noted that the degree of genetic differentiations does not always equate to dispersal abilities among species and is often affected by population history (e.g.16). Future studies of movement patterns, either by swimming or by passive larval dispersal, would be useful to explain the population structures we identified in this study.
In this study, we mainly used COI sequences to clarify basic information on the genetic population structures of organisms near hydrothermal vent fields. It is important, however, to be cautious when interpreting our results. While there have been numerous studies that have based their results and conclusions on COI sequences including for deep-sea species11,16, the genetic marker we chose was not as variable as those commonly used for coastal marine organisms54. In future studies, analyses using highly variable genetic markers such as microsatellites55 and single nucleotide polymorphisms (e.g. MIG-seq used in this study) should be used to detect more detailed population structures. Highly polymorphic markers have already been used in deep-sea amphipods55,56, and it is hoped that similar analyses will be applied to near-vent amphipods from this study in the future. Such information on genetic connectivity would be essential in assessing the environmental impacts of mineral mining around hydrothermal vents, as well as to determine potential locations of deep-sea marine protected areas57,58. Although a trial has been conducted to infer community resilience in hydrothermal vents considering a dispersal network10, the connectivity data supporting this dispersal network is still limited, thus it is necessary to accumulate connectivity data for hydrothermal vents and their surrounding ecosystems in the future.
Data availability
All sequences and fastq files were deposited in DNA Data Bank of Japan database (LC532844-LC533073, DRA014289).
References
Van Dover, C. L. et al. Environmental management of deep-sea chemosynthetic ecosystems: justification of and considerations for a spatially based approach. ISA Technical Study: No.9. (International Seabed Authority, 2011).
Ikehata, K., Suzuki, R., Shimada, K., Ishibashi, J., & Urabe, T. Mineralogical and Geochemical Characteristics of Hydrothermal Minerals Collected from Hydrothermal Vent Fields in the Southern Mariana Spreading Center. In Subseafloor biosphere linked to hydrothermal systems: TAIGA Concept. 275–288 (Springer Tokyo, 2015).
Rona, P. A. & Scott, S. D. A special issue on sea-floor hydrothermal mineralization; new perspectives; preface. Econ. Geol. 88, 1935–1976 (1993).
Glasby, G. P., Iizasa, K., Yuasa, M. & Usui, A. Submarine hydrothermal mineralization on the Izu-Bonin arc, south of Japan: an overview. Mar. Georesources Geotech. 18, 141–176 (2000).
Van Dover, C. L. Inactive sulfide ecosystems in the deep sea: a review. Front. Mar. Sci. 6, 461. https://doi.org/10.3389/fmars.2019.00461 (2019).
Boschen, R. E., Rowde, A. A., Clark, M. R. & Gardner, J. P. Mining of deep-sea seafloor massive sulfides: a review of the deposits, their benthic communities, impacts from mining, regulatory frameworks and management strategies. Ocean Coast. Manag. 84, 54–67 (2013).
Washburn, T. W. et al. Ecological risk assessment for deep-sea mining. Ocean Coast. Manag. 176, 24–39 (2019).
Matsui, T., Sugishima, H., Okamoto, N., Igarashi, Y. Evaluation of turbidity and resedimentation through seafloor disturbance experiments for assessment of environmental impacts associated with exploitation of seafloor massive sulfides mining. Proceedings of the Twenty-eighth. International Ocean and Polar Engineering Conference. 144–151 (2018).
International Seabed Authority. Recommendations for the guidance of contractors for the assessment of the possible environmental impacts arising from exploration for marine minerals in the Area. https://www.isa.org.jm/documents/isba19ltc8 (2013).
Suzuki, K., Yoshida, K., Watanabe, H. & Yamamoto, H. Mapping the resilience of chemosynthetic communities in hydrothermal vent fields. Sci. Rep. 8, 9364. https://doi.org/10.1038/s41598-018-27596-7 (2018).
Yahagi, T., Watanabe, H., Ishibashi, J. I. & Kojima, S. Genetic population structure of four hydrothermal vent shrimp species (Alvinocarididae) in the Okinawa Trough, Northwest Pacific. Mar. Ecol. Prog. Ser. 529, 159–169 (2015).
Mullineaux, L. S. Deep-sea hydrothermal vent communities. In Marine community ecology and conservation (eds Bertness, M. D. et al.) 383–400 (Sinauer, 2013).
Van Dover, C. L., German, C. R., Speer, K. G., Parson, L. M. & Vrijenhoek, R. C. Evolution and biogeography of deep-sea vent and seep invertebrates. Science 295, 1253–1257 (2002).
Yahagi, T., Kayama-Watanabe, H., Kojima, S. & Kano, Y. Do larvae from deep-sea hydrothermal vents disperse in surface waters?. Ecology 98, 1524–1534 (2017).
Hebert, P. D. & Gregory, T. R. The promise of DNA barcoding for taxonomy. Syst. Biol. 54, 852–859 (2005).
Iguchi, A. et al. Comparative analysis on the genetic population structures of the deep-sea whelks Buccinum tsubai and Neptunea constricta in the Sea of Japan. Mar. Biol. 151, 31–39 (2007).
Goode, G. B. & Bean, T. H. A catalogue of the fishes of Essex County, Massachusetts, including the fauna of Massachusetts Bay and the contiguous deep waters. Bull. Essex Inst. 11, 1–38 (1879).
Johnson, J. Y. Descriptions of some new genera and species of fishes obtained at Madeira. Proc. Zool. Soc. Lond. 1862, 167–180 (1862).
Bate, C. S. Report on the Crustacea Macrura collected by the Challenger during the years 1873–76. Report on the scientific results of the Voyage of H.M.S. Challenger during the years 1873–76. Zoology 24, 1–942 (1888).
Folmer, O., Black, M., Hoeh, W. R., Lutz, R. & Vrijenhoek, R. C. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol Biotech. 3, 294–299 (1994).
Pilgrim, E. M., Blum, M. J., Reusser, D. A., Lee, H. & Darling, J. A. Geographic range and structure of cryptic genetic diversity among Pacific North American populations of the non-native amphipod Grandidierella japonica. Biol. Invasions 15, 2415–2428 (2013).
Suyama, Y. & Matsuki, Y. MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5, 16963. https://doi.org/10.1038/srep16963 (2015).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2020).
Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 11, e0163962. https://doi.org/10.1371/journal.pone.0163962 (2016).
Paradis, E. pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420 (2010).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Darriba, D. et al. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2020).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RaxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Ronquist, F. R. & Huelsenbeck, J. P. MRBAYES 3: Bayesian inference of phylogeny. Bioinformatics 19, 1572–1574 (2003).
Puillandre, N., Brouillet, S. & Achaz, G. ASAP: assemble species by automatic partitioning. Mol. Ecol. Resour. 21, 609–620 (2021).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, http://journal.embnet.org/index.php/embnetjournal/article/view/200/479 (2011).
Rochette, N. C., Rivera-Colón, A. G. & Catchen, J. M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754 (2019).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186 (2013).
Oksanen, J. et al. vegan: Community Ecology Package. R package version 2.5–6. https://CRAN.R-project.org/package=vegan (2019).
Dana, J. D. Synopsis of the genera of Gammaracea. Am. J. Sci. Arts 8, 135–140 (1849).
Hansen, H. J. Malacostraca marina Groenlandiæ occidentalis Oversigt over det vestlige Grønlands Fauna af malakostrake Havkrebsdyr. Vidensk. Meddel. Natuirist. Foren Kjobenhavn, Aaret 9, 5–226 (1888).
Van Dover, C. L. The ecology of deep-sea hydrothermal vents (Princeton University Press, 2000).
Tunnicliffe, V. The biology of hydrothermal vents: ecology and evolution. Oceanogr. Mar. Biol. Annu. Rev. 29, 319–407 (1991).
Priede, I. G., Bagley, P. M., Smith, A., Creasey, S. & Merrett, N. R. Scavenging deep demersal fishes of the Porcupine Seabight, north-east Atlantic: observations by baited camera, trap and trawl. J. Mar. Biol. Assoc. U. K. 74, 481–498 (1994).
Causse, R., Biscoito, M. & Briand, P. First record of the deep-sea eel Ilyophis saldanhai (Synaphobranchidae, Anguilliformes) from the Pacific Ocean. Cybium 29, 413–416 (2005).
King, N. J., Bagley, P. M. & Priede, I. G. Depth zonation and latitudinal distribution of deep-sea scavenging demersal fishes of the Mid-Atlantic Ridge, 42 to 53°N. Mar. Ecol. Prog. Ser. 319, 263–274 (2006).
Leitner, A. B., Durden, J. M., Smith, C. R., Klingberg, E. D. & Drazen, J. C. Synaphobranchid eel swarms on abyssal seamounts: largest aggregation of fishes ever observed at abyssal depths. Deep Sea Res. Oceanogr. Res. Part I Pap. 167, 103423. https://doi.org/10.1016/j.dsr.2020.103423 (2021).
Fishelson, L. Comparative internal morphology of deep-sea eels, with particular emphasis on gonads and gut structure. J. Fish. Biol. 44, 75–101 (1994).
Bailey, D. M. et al. High swimming and metabolic activity in the deep-sea eel Synaphobranchus kaupii revealed by integrated in situ and in vitro measurements. Physiol. Biochem. Zool. 78, 335–346 (2005).
Trenkel, V. M. & Lorance, P. Estimating Synaphobranchus kaupii densities: contribution of fish behaviour to differences between bait experiments and visual strip transects. Deep Sea Res. Oceanogr. Res. Part I Pap. 58, 63–71 (2011).
Raupach, M. J. et al. Genetic homogeneity and circum-Antarctic distribution of two benthic shrimp species of the Southern Ocean, Chorismus antarcticus and Nematocarcinus lanceopes. Mar. Biol. 157, 1783–1797 (2010).
Dambach, J., Raupach, M. J., Leese, F., Schwarzer, J. & Engler, J. O. Ocean currents determine functional connectivity in an Antarctic deep-sea shrimp. Mar. Ecol. 37, 1336–1344 (2016).
Dambach, J., Raupach, M. J., Mayer, C., Schwarzer, J. & Leese, F. Isolation and characterization of nine polymorphic microsatellite markers for the deep-sea shrimp Nematocarcinus lanceopes (Crustacea: Decapoda: Caridea). BMC Res. Notes 6, 75. https://doi.org/10.1186/1756-0500-6-75 (2013).
Ritchie, H., Jamieson, A. J. & Piertney, S. B. Phylogenetic relationships among hadal amphipods of the Superfamily Lysianassoidea: Implications for taxonomy and biogeography. Deep Sea Res. Part I 105, 119–131 (2015).
Bowen, B. W. et al. Phylogeography unplugged: comparative surveys in the genomic era. Bull. Mar. Sci. 90, 13–46 (2014).
Ritchie, H., Jamieson, A. J. & Piertney, S. B. Population genetic structure of two congeneric deep-sea amphipod species from geographically isolated hadal trenches in the Pacific Ocean. Deep Sea Res. Part I. 119, 50–57 (2017).
Iguchi, A. et al. Deep-sea amphipods around cobalt-rich ferromanganese crusts: taxonomic diversity and selection of candidate species for connectivity analysis. PLoS ONE 15, e0228483. https://doi.org/10.1371/journal.pone.0228483 (2020).
Baco, A. R. et al. A synthesis of genetic connectivity in deep-sea fauna and implications for marine reserve design. Mol. Ecol. 25, 3276–3298 (2016).
Taylor, M. L. & Roterman, C. N. Invertebrate population genetics across Earth’s largest habitat: the deep-sea floor. Mol. Ecol. 26, 4872–4896 (2017).
Acknowledgements
We thank M. Kojima for her technical help. We also thank S. Kato and T. Matsui of JOGMEC for their assistance in implementing the project. This study was commissioned by Agency for Natural Resources and Energy, Ministry of Economy, Trade and Industry of Japan. Additional support was provided by the Research Laboratory on Environmentally-conscious Developments and Technologies (E-code) at the National Institute of Advanced Industrial Science and Technology (AIST).
Author information
Authors and Affiliations
Contributions
H.K. and A.I. designed the experiments on the basis of the specimens collected by N.O. T.I., Y.O., K.G., and M.N. sorted samples and performed molecular experiments. H.K., A.I. and M.N. analyzed the data. A.S., Y.T., and N.O. contributed to the materials and reagents. H.K., A.I., T.W., and T.K. wrote the main text of the manuscript. All authors contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that this study received funding from JOGMEC. At the time of research, TI, YO, and KG were employed by KANSO Technos CO.,LTD., and NO was employed by JOGMEC. JOGMEC and KANSO Technos CO.,LTD. did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The remaining authors, HK, AI, YT, TWW, MN, TK, and AS declare no additional competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kise, H., Iguchi, A., Ikegami, T. et al. Genetic population structures of common scavenging species near hydrothermal vents in the Okinawa Trough. Sci Rep 13, 2348 (2023). https://doi.org/10.1038/s41598-022-14100-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14100-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
