
Abstract
Fe(III) 5,10,15,20-(tetraphenyl)porphyrin chloride (FeTPP) and Co(III) 5,10,15,20-(tetraphenyl)porphyrin chloride (CoTPP) were adsorbed on carbon Vulcan and studied as electrocatalysts for the oxygen reduction reaction (ORR) before and after pyrolysis. The pyrolysis process was also simulated through ab initio molecular dynamic simulations and the minimum energy path for the O2 dissociation after the interaction with the metal center of the FeTPP and CoTPP were calculated. After the pyrolysis the FeTPP showed the best performances reducing O2 completely to H2O with increased limiting current and lower overpotential. Tafel slops for the various catalysts did not change after the pyrolytic process suggesting that the mechanism for the ORR is not affected by the heat treatment. TEM images, X-ray diffraction, XPS spectroscopy, 57Fe Mössbauer, and DFT simulations, suggest that there is no breakdown of the macrocyclic complex at elevated temperatures, and that the macro cyclic geometry is preserved. Small variations in the Metal-O2 (M-O2) binding energies and the M–N bond length were observed which is attributed to the dispersive interaction between the macrocycles and the irregular surface of the Vulcan substrate induced by the heat treatment and causing better interaction with the O2 molecule. The theoretical strategy herein applied well simulate and explain the nature of the M–N–C active sites and the performances towards the ORR.
Similar content being viewed by others

Introduction
Several plans of action are under study to decrease the environmental burden caused by the accumulation of greenhouse gases1. Low-temperature fuel cells emerge as a promising strategy to this proposal2,3. The H2 oxidation at the anode and the O2 reduction reaction (ORR) at the cathode would allow to secure up to four-electrons with a theoretical potential of 1.23 V and to obtain water as by-product. The cathodic reaction is the rate limiting step which is hampering the mass diffusion of this technology because it is too slow in alkaline media and unstable at low pHs. Platinum and Pt-based catalysts accelerate the reaction to appreciable paces4,5, but the price and scarcity of this noble metal are the limits6. Transition metals nitrogen complexes (MN4) have been studied as a possible replacement for the Pt-metal group since 1964, when Jasinsky reported that Co phthalocyanine (CoMN4) had catalytic activity for the ORR7. But MN4 complexes show lower performances than the Pt catalysts and no-stability in acid8,9,10. To increase the catalytic activities several strategies have been devised. Among them: (i) the use of carbon-based nanomaterial as supporting matrix to increase surface area, and conductivity11,12,13,14,15; (ii) the shift of the metal redox center (Mn+1/n) to more positive values with electron-withdrawing peripherical substituents, so to increase the onset potential (Eonset) for the ORR according to volcano correlation10,16,17,18,19; (iii) the pyrolysis of MN4 to modify the N-metal bond length (M–N–C) and the interaction of the metal center with O220,21,22,23,24,25 (M-O2). Jahnke and co-workers were the pioneers of pyrolyzing complexes, with a seminal work in 197626. Later developments have seen the use of metal-salts (Co, Fe, Mn) and N–C matrices as independent precursors to obtain active pyrolyzed catalysts for the ORR27,28,29,30,31,32,33,34,35.
After heat treatment of MN4 catalysts, the identification of the M–N–C active site is a crucial step to obtain insights into the ORR mechanism. Spectroscopic technics as Mössbauer, XPS, EXAFS, and XAS20,24,36,37,38,39,40,41, are powerful tools to define the M-Nx coordination nature. Density functional theory (DFT) calculations have been developed to study the interactions between the N atoms and the metal centers of the catalyst and to obtain the binding energy (Ebin) as thermodynamic descriptor for the ORR42,43,44. When integrated with spectroscopic and electrochemical experiments, DFT simulations, can merge experimental results granting comprehensive perspectives32,45,46. It is a general agreement that MNx, and MNx+1 (where x: 2 + 2 or 4, and M is Co or Fe) are the most M–N–C active sites for the ORR where the formation is the result of the insertion of the metal into the carbon matrix28,37,38,47,48. However, when the MN4 complex is supported on carbon matrix, and then pyrolyzed, the nature of the active site is still under debate. Some authors claim that the macrocycle is broken up at low pyrolysis temperatures, (~ 800 °C, which is also considered as the optimal pyrolysis temperature to obtain electrocatalysts49,50,51), and then the MN4 active site is inserted into the carbon matrix, analogously to others M–N–C formed by independent precursors21,24,52. Other research groups suggest that the macrocyclic structures do not change after the heat treatment and remain adsorbed on the carbon support or that only the peripheral substituents of the MN4 are removed53,54,55.
To obtain more insights into the electrocatalytic active sites of pyrolyzed MN4 complexes, we performed ab initio molecular dynamics to simulate the pyrolysis process of Fe(III) 5,10,15,20-(tetraphenyl)porphyrin chloride (FeTPP) and Co(III) 5,10,15,20-(tetraphenyl)porphyrin chloride (CoTPP). We correlated the simulated results to spectroscopic (TEM images, X-ray diffraction, XPS spectroscopy, 57Fe Mossbauer) and electrochemical data. The calculation methodology is proposed to study the formation of M–N–C active sites, from the beginning of the thermal process, without limiting the calculations to a specific metal active site56,57,58,59.
Theoretical studies
Co and Fe tetraphenyl porphyrin (CoTPP and FeTPP) were selected as catalysts for the ORR because of their strong \(\pi\)–\(\pi\) stacking interactions with the carbonaceous matrix (carbon Vulcan), used as substrate, and for the good electrocatalytic activity60,61,62,63,64. The band structure calculation shows that the amorphous carbon has metallic characteristic, as shown in supporting information Fig. S1. Figure 1a shows the equilibrium geometry of the FeTPP adsorbed on the amorphous carbon (a-C) after pyrolysis at 800 °C and subsequent optimization. The FeTPP molecule is strongly physiosorbed on the irregular a-C surface with binding energy of − 2.27 eV. The system shows a magnetic moment m = 2.24 μB. Similar results were found for the CoTPP molecule on a-C, but with binding energy and magnetic moment of − 2.25 eV and 1.00 μB, respectively. The molecules tend to preserve their spin multiplicity after the adsorption on the a-C substrate. However, it is observed a small spin polarization of the substrate resulting from a net magnetization higher than the isolated molecule, which are found of 0.86 and 2.06 μB for CoTPP and FeTPP, respectively.

(a) Equilibrium geometry of the FeTPP molecule adsorbed on amorphous carbon after heat treatment. (b) Minimum energy path for the O2 dissociation after the interaction with the metal center of the FeTPP and CoTPP which are supported on a-C (which is removed from de inset figures for clarity). The initial, saddle point and final geometries of O2 interacting with FeTPP through the reaction coordinate are also shown.
The O2 interaction with the metal center of FeTPP and CoTPP supported on a-C were investigated. O2 attaches on Fe and Co atoms with binding energies of − 0.64 and − 0.48 eV, respectively, following the end-on configurations. A small reduction in the O2 binding energy by the action of the carbon substrate can be noticed. Details of the O2 attachment on the porphyrins are shown in Table 1. The spin coupling between O2 and the metal atoms results in a net magnetization of 0.80 and 0.72 μB for FeTPP and CoTPP, indicating a low spin configuration of the adducts. The O–O bond distance of the adsorbed O2 increases by around 7% on the Fe and 6% on the Co, with respect to gas phase O2, suggesting an ORR catalytic activity. To verify this activity, we calculated the minimum energy path and saddle point for the O2 dissociation starting from initial and final geometries previously optimized, using the NEB method. The results are shown in Fig. 1b. An energy barrier of 1.15 eV for the O2 dissociation after the interaction with the metal center of the carbon-supported FeTPP, was calculated as shown in the inset images of Fig. 1b. Similar results are found for the O2 dissociation on the carbon-supported CoTPP, but on this macrocycle, the energy barrier is much higher (1.42 eV). The results reveal a much higher ORR activity for the FeTPP than CoTPP, which is also better than those found on Fe Phthalocyanine supported on carbon nanotubes without pyrolysis process and previously reported14,62.
In order to verify the stability of the FeTPP macrocycle on a-C at higher pyrolysis temperature, we performed AIMD simulation at 1500 °C, during 2 ps of simulation time. The FeTPP structural stability is preserved during the simulation time, revealing the stiffness of the macrocycle. A snapshot of the a-C:FeTPP geometry taken at 2 ps of the AIMD simulation is shown in supporting information Fig. S2.
Spectroscopic studies
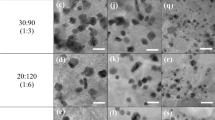
In Fig. 2a–d we show the TEM images of the surface structure of the supporting carbon material before (Fig. 2a,c) and after (Fig. 2b,d) heat treatment. After pyrolysis a more exposed structure can be observed. Nonetheless, major structural changes are excluded. Along with this, we could not find presence of nanoparticles or crystals formed by π-π stacking65,66,67. This fact is also supported by X-ray diffraction (Fig. S3), where samples at 25 °C, as well as 800 °C show a characteristic peak at approximately 2θ = 25.4° and 43.3° associated with graphite (002) and (100) planes for attached MN4 complex68,69,70. Also, no nanoparticles diffraction peak is visualized before or after heat treatment, suggesting that the possible variation in electrocatalytic performances should not be attributed to the formation of nanoparticles or nitrides and just to MN4 sites16,22,71,72,73,74.

Transmission electron microscopy images of (a) FeTPP 25 °C, (b) FeTPP 800 °C, (c) CoTPP 25 °C, and (d) CoTPP 800 °C adsorbed on Vulcan XC-72.
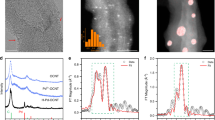
The samples were also characterized by XPS spectroscopy (Fig. 3a,b and Fig. S4a,b) using procedures similar to those used in previous works22,71. Figure 3a,b show the N 1 s spectra recorded from FeTPP (25 °C and 800 °C) and CoTPP (25 °C and 800 °C). The load of iron in the FeTPP catalysts was quite small and this precluded recording Fe 2p XPS spectra with good statistics. The spectra were computer-fitted using the constraint of equal full width at half maximum (FWHM) for all the spectral components in order to avoid undesired fluctuations in the relative obtained areas. Five contributions were used to fit adequately the experimental data which were located at the binding energies 403.9 eV (N-oxide) 402.1 eV (N-graphitic), 400.9 eV (N-pyrrolic), 398.5 eV (N-pyridinic), and, finally, at 399.9 eV (MN4 sites; M: Fe or Co) and which are usually associated in the literature with the catalytic M–N–C active site16,22,71,72,73. The relative areas of the different components and their assignment to the various chemical species are collected in Table S1 of the supplementary information. The spectra recorded from the Fe- and CoTPP 25 °C are both dominated by the MN4 contribution (Fig. 3a,b). After pyrolysis at 800 °C the intensity of this component is reduced significantly in both the iron- and cobalt materials. This might be related with a smaller concentration of iron and cobalt (vide infra) in the pyrolyzed samples as compared with the original samples which is consequently reflected in a smaller number of MN4 sites. It must be noted that the N 1 s spectra of the pyrolyzed materials have a lower signal-to-noise ratio than the corresponding of the 25 °C samples. Therefore, it seems that the pyrolysis treatment also reduces the nitrogen content of the materials. It is quite remarkable that the iron and cobalt samples show only small differences in their respective N 1 s spectra. This suggests that the preparation method of the materials before and after pyrolysis is quite reliable and the behavior almost independent of the type of metal.

(a,b) N1s XPS spectra recorded from the FeTTP and CoTPP materials adsorbed on Vulcan XC-72 before and after pyrolysis; (c) room temperature 57Fe Mossbauer spectra recorded from the FeTPP materials; (d) Co 2p XPS spectra recorded from the CoTPP materials.
To gather more information about the nature of the iron species in these samples, we resorted to Mossbauer spectroscopy (Fig. 3c). The Mossbauer spectra also showed a poor signal-to-noise ratio as a consequence of a small iron load. The absorption of the spectrum corresponding to the FeTPP 800 °C sample is much lower than that shown by the FeTPP 25 °C sample, indicating that the latter has a larger iron concentration. This supports the reduction observed in the intensity of the Fe-N4 signal in the N 1 s XPS spectra. The room temperature 57Fe Mössbauer spectrum recorded from FeTPP 25 °C consisted of a single, narrow paramagnetic doublet with hyperfine parameters δ = 0.38 mms-1, Δ = 1.18 mms−1. These parameters are characteristic of low spin Fe(II) in FeIIN4 species39. The result is important since (i) it shows the presence of this specie in the freshly prepared catalyst confirming the XPS results and (ii) it indicates that this is the only iron species present in this material. The Mossbauer spectrum recorded from the FeTPP 800 °C sample (Fig. 3c) is different. It shows two different paramagnetic doublets. The minor one (27% of the spectral area) has identical parameters than the doublet present in the spectrum of FeTPP 25 °C and, therefore, it corresponds to FeIIN4 sites. The hyperfine parameters of the major doublet (77%) δ = 0.14 mms−1, Δ = 0.51 mms−1 cannot be associated to any of the usual iron species appearing in this type of materials39 and resemble more those shown by some defective paramagnetic cubic iron mononitrides such as \(\upgamma \prime \prime \prime\)-FeN75. Such type of nitrides can be stabilized by rapid quenching after heating above 800 °C and, being of austenic nature, where the presence of carbon might help in this stabilization.
The Co 2p spectra from the cobalt-containing materials are shown in Fig. 3d. The signal-to-noise ratio of the Co 2p spectrum corresponding to CoTPP 800 °C sample was smaller than that of CoTPP 25 °C. This, again, suggests a smaller metal content in the pyrolyzed materials. The Co 2p spectrum recorded from CoTPP 25 °C consists of a spin orbit doublet with intense shake-up structure behind the main Co 2p3/2 and Co 2p1/2 photoemission lines. The binding energies corresponding to these core levels are 780.7 eV (Co 2p3/2) and 796.4 eV (Co 2p1/2) while the binding energies of the shake-up satellites are 785.8 eV and 803.0 eV. The structure of the spectrum and the binding energies of the main core level lines, with a spin–orbit energy separation of 15.7 eV, are characteristic of a high spin S = 3/2 Co(II) species76,77. The Co 2p spectrum recorded from the CoTPP 800 °C, looks different. It has a less prominent shake up structure appearing at 786.5 eV and 799.8 eV (the latter satellite being very weak), a multiplet splitting component at 783.6 eV and main photoemission lines appearing at the binding energies of 780.5 eV (Co 2p3/2) and 795.2 eV (Co 2p3/2), i.e., with a spin–orbit splitting energy of 14.7 eV (1 eV smaller than in the other sample). All these data together can be associated with the presence of an intermediate spin S = 1 Co(III) species13.
Resuming, the spectroscopic results show that the amorphous carbon structure is not significantly altered by the heat treatment and that the FeTPP and the CoTPP are mainly incorporated to the carbon support by strong π-π stacking with an N-M bonds length which might become slightly larger after pyrolysis as in accordance to the DFT calculations (Table 1) and to a more exposed structure as observed in TEM images (Fig. 2). The concentration of Fe and Co decreases during the pyrolysis process accompanied with the formation of nitrides.
Electrochemical studies
Figure 4a,b shows cyclic voltammetry in N2 atmosphere together with their respective linear sweep voltammetry (LSV, at 1600 rpm) curves in O2 saturated buffer for pyrolyzed and unpyrolyzed FeTPP and CoTPP, and Vulcan XC-72 in alkaline media, Fig. 4c. All the obtained values are summarized in Table 2. To determine the Tafel plot in Fig. 4d, the LSV was corrected by the limiting current contribution [jL* j]/[jL-j]78 before using Butler–Volmer equation78 and to avoid diffusional contribution (jL) in the kinetic electron-transfer process (jk). The M(III)/(II) formal potential has been detected by cyclic voltammetry and is shown with a grey circle in Fig. 4a,b. The values are in agreement with those reported in the literature16,22,71. As reported in Table 2 and Fig. 4a,b, and graphical abstract, there is a connection between the formal potential of the M(III)/(II) redox couple and the onset potential (Eonset) for the ORR, and an increase in the catalytic current79. This can be noticed for all the catalysts (i.e., before and after the heat treatment) and has been widely reported for monolayers of porphyrins and phthalocyanines as well as for pyrolyzed complexes10,16,80. Further, from the electrochemical results, two facts can be observed (i) for non-pyrolyzed Co and Fe porphyrins, the CoTPP shows higher catalytic activity than FeTPP (Fig. 4c, dash black and red line respectively) in agreement with the lower ionization potential of Fe-porphyrins which make them worse catalysts than cobalt counterpart16,81,82,83. (ii) There is a shift of E0’ for Co(III)/Co(II) and Fe(III)/Fe(II) to more positive values after pyrolysis, which indicates an environmental electronic modification that affects the metal redox centers and the catalytic activity, where the M(II) sites are required for the ORR process to take place in basic media as for an inner-sphere electron transfer mechanism (ISET)71,84,85. Several authors affirm that changes in the nature of carbonaceous support matrix as graphitic/amorphous structure or surface defects after the pyrolysis could be responsible for changes in catalytic activity toward the ORR with intrinsic modification in spin distribution for the carbon matrix86,87,88,89. This is in agreement with the herein reported DFT simulations. The electrons transferred during the ORR were calculated with the rotating ring-disk electrode (Eq. (3))78. Results are resumed in Table 2 and show numbers close to 3 and 4 electrons transferred by CoTPP and FeTPP respectively. A lower percentage of hydrogen peroxide is produced by FeTPP, before and after heat treatment, compared to CoTPP in similar conditions (values are resumed in Table 2). These results indicated direct H2O formation or 2 + 2 mechanism for FeTPP90, while for CoTPP water and peroxide formation are simultaneously formed during the reduction mechanism. FeTPP, after pyrolysis, showed better performance for the ORR than CoTPP, as we expected from DFT calculations of the energy barrier for the O2 dissociation in Fig. 1b, because of the lower interplay between O2 and Co metal center in contrast to Fe centers (see Table 1). In Fig. 4d, similar Tafel slope values for Co and Fe porphyrins before and after pyrolysis are reported. The value is close to − 0.060 V dec−1 at lower overpotential, where M(II) concentrations are depending on the applied potential22,91. The Tafel slope value indicates that the rate-determining step (r.d.s.) is chemical O2 adsorption in M(II) preceded by a fast first electron transfer for ISET mechanism, which is a common step in the studied catalysts22,87,92 according to the following equation:

Cyclic voltammetry at 100 mV s−1 in N2 saturated atmosphere (black line) and polarization curve iL corrected [(iL*i)/(iL-i)] in O2 saturated atmosphere (red line) with 0.1 M NaOH as electrolytic solution for (a) FeTPP, FeTPP-800 °C, and Vulcan XC-72 (b) CoTPP, CoTPP-800 °C, and Vulcan XC-72. (c) Ring-disk current curves for linear sweep voltammetry at 5 mV s−1 in 0.1 M NaOH O2 saturated atmosphere for FeTPP or CoTPP at 25(dash line) and after pyrolysis at 800 °C (CoTPP-800 °C, FeTPP-800 °C, solid line), and commercial Pt/C 20 wt% (gray dash line) as from Fig. S5. (d) Tafel plot for the ORR for FeTPP and CoTPP before and after pyrolysis at 800 °C.
In light of these results (values resumed in Table 2), there is no significant variation in the ORR mechanism before and after heat treatment, suggesting the similar nature of the M–N–C active site after pyrolysis. Nevertheless, the structure of the resulting catalysts is enormously depending on the synthesis route and pyrolysis process93,94,95,96. We attribute the enhancement in the catalytic activity for the ORR to variations in the interactions with the Vulcan substrate, which induces fluctuations in the electron density of the metal redox center, causing changes to the binding energy between M-O2 as reported in Table 1. This also relates with the differences in the M–N bond length in the catalyst structure after pyrolysis. Finally, if we consider the catalytic activity as log jk at constant potential (E: 0.8 V vs. RHE) versus binding energy M-O2 and also versus the formal potential, as proposed by several authors10,16,61,97,98,99, we obtain the figure reported as graphical abstract, where the MN4 structure is maintained, and the catalysts follow the trend of the correlations.
Conclusions
Ab initio molecular dynamic simulations were carried out for the pyrolysis of CoTTP and FeTPP adsorbed on amorphous carbon. The theoretical results indicated that there is no breakdown of the macrocyclic complex at elevated temperatures, up to 1500 °C, preserving the MN4 geometry. However, variations in the M-O2 binding energies were observed after the simulating pyrolysis at 800 °C, which is attributed to the dispersive interaction between the macrocycles and the irregular surface of the a-C substrate, inducing small variation in the MN4 bond distance. TEM images showed a more exposed structure after the pyrolytic process. XRD measurements excluded the presence of nanoparticles or MN4 stacks or crystals before or after the pyrolysis. The XPS results corroborate the presence of MNx catalytic sites for both complexes, before and after heat treatment. Through electrochemical studies we could detect a displacement of the formal potential to more positive values after the thermal treatment, but the Tafel slopes did not vary showing that the ORR mechanism did not change before and after the pyrolysis. It is suggested that the variation in the catalytic activity for the ORR is associated with the variations in the interaction of Fe and Co porphyrins with the carbon substrate, where the porphyrinic M–N–C active site is preserved after pyrolysis. If it is desired to introduce nitrogen and metal atoms into the carbon structure, other strategies have to be adopted, such as the solid-state synthesis with metal salts and carbon–nitrogen sources instead of using metallo-macro-cycles. The DFT calculations proved to be a powerful tool to simulate the changes of M–N–C active sites during pyrolytic processes.
Materials and methods
Catalysts preparation and synthesis
Fe(III) 5,10,15,20-(tetraphenyl)porphyrin chloride and Co(III) 5,10,15,20-(tetraphenyl)porphyrin chloride were supplied by PorphyChem (Dijon, France). Both metalloporphyrins were mixed separately with Vulcan XC-72® in agata mortar for 40 min (MN4:XC-72; 1:4 mass ratio), then 1 mg of catalyst powder was dispersed in 1 mL of isopropanol: water (1:4). The resulting ink was stirred for 5 h, then dried at 60 °C for 5 h. The first fraction of obtained powder was denoted as FeTPP-25 °C and CoTPP-25 °C. A second fraction was pyrolyzed under N2 atmosphere at 800 °C, carried out in a Furnace 1200 °C (SK-G03123K). The pyrolyzed samples were labelled FeTPP-800 °C and CoTPP-800 °C, respectively. Finally, the working electrode was modified by a spin coating process, dropping 10 µL of ink (Co or FeTTP at 25 or 800 °C) containing 1 mg of powder catalyst in 1 mL of isopropanol:water, 1:4, and 0.7 Vol% of 5% Nafion® solution, to final surface concentration of 0.05 mg cm−2 of the catalysts. Pt/C 20 wt% and Vulcan XC-72 (FuellCell) ink was obtained using a similar procedure as for the FeTPP-25 °C and CoTPP-25 °C catalysts.
Theoretical simulations
Spin-polarized density functional theory (DFT) calculations were performed using the Quantum Espresso ab initio package100. Dispersive interactions were included by the non-local van der Waals correlations into the exchange correlation potential (vdW-DF2)101,102,103,104,105. Kohn–Sham eigenfunctions are expanded on a plane-waves basis set where the interaction between valence electrons and ion cores are described by ultrasoft pseudopotentials106. Converged results have been achieved by cut off energies of 480 eV. All atoms are left free to relax until the residual force on each atomic component was less than 0.002 eV/Å. The sampling of the Brillouin zone was performed with the Γ point. The minimum energy path for the O2 dissociation was obtained using the climbing Nudged Elastic Band (NEB) method107,108, using 8 images throughout the reaction coordinate. Amorphous carbon was obtained from the diamond structure through ab initio molecular dynamic (AIMD) simulation, using the Vienna Ab Initio Simulation Package (VASP)109. Here we employ the generalized gradient approximation for the exchange–correlation functional and a cut off energy of 400 eV for the plane-wave basis set. The core-valence interaction was described by the projector augmented-wave method110. A large periodic unit cell of 17.8 × 17.8 × 16.0 Å3 containing a diamond slab with 7 monolayers was thermalized at 4000 K during a simulation time of 5 ps and then cooled down to 0 K during 2 ps, using a time step of 1 fs. The resulting structure was then optimized by self-consistent DFT calculations after the equilibrium geometry is achieved. Pyrolyzed FeTPP and CoTPP adsorbed on the amorphous carbon structure was obtained by AIMD simulation. The system was thermalized at 1073 K during a simulation time of 2 ps and then cooled down to 0 K during 2 ps.
Spectroscopic characterization
The transmission electronic microscopy (TEM) was performed by depositing the ink on the TEM grid until it dried. The TEM images were obtained using Hitachi HT7700 equipment with an accelerating voltage of 120 kV.
X-ray diffraction (XRD) was used to determine the nanoparticle presence before and after-heat treatment, performed in Bruker D8 Advance diffractometer with CuKα1,2 radiation (λ = 1.54118 Å) at 40 mA and 40 kV.
X-ray photoelectron spectroscopy data were recorded under a vacuum better than 2.0 × 10–9 mbar using a PHOIBOS-150 electron analyzer (SPECS), with a constant pass energy of 20 eV and Al Kα radiation. The binding energy scale was referenced to the main C–C C1s signal located at 284.6 eV.
57Fe Mossbauer spectra were recorded from the iron-containing samples. The data were taken in the transmission mode at room temperature using a conventional constant acceleration spectrometer. The velocity scale was calibrated using a natural alpha-iron foil 6 μm thick. The spectra were computer-fitted and the isomer shifts referred to the centroid of the sextet of alpha-iron at room temperature.
Electrochemical characterization
A typical three electrodes electrochemical cell was used during the electrochemical experiments, using Hg0/Hg2Cl2 calomel electrode and carbon stick as reference and counter electrode respectively. Edge-plane pyrolytic graphite disk mounted into a Teflon rotating shaft containing the Pt0 ring as working electrode. The disk was 5 mm diameter and 4 mm thick, while the ring diameter was 6.5 mm and the external diameter of the ring was 7.50 mm. The rotor system for the -ring-disk electrode was from PINE Instrument (Durham, NC, USA). Sodium hydroxide from Merck Millipore (NaOH, ≥ 99%) was used to prepare 0.1 M NaOH as electrolytic solution with Milli-Q water (18.2 MΩ·cm at 25 °C). Cyclic voltammetry characterization was done at 100 mV s−1 to observe faradic processes using 0.1 M NaOH electrolytic solution and N2 saturated atmosphere. The electrocatalytic activity was studied at 5 mV s−1 in O2 saturated atmosphere, using an Autolab PGSTAT 204 bipotentiostat. The electrons transferred and the peroxide formation percentage during the ORR was determined by rotating -ring -disk using Eqs. (3) and (4) respectively78,111,112:
where IR = ring current, ID = ring current; N = collection efficiency (0.39). Finally, Tafel slopes were obtained by Butler–Volmer equation78 to derivate the determining rate step in each catalyst.
Data availability
The data generated and analysed during the current study are available from the corresponding author.
References
Senjyu, T. & Howlader, A. M. Operational aspects of distribution systems with massive DER penetrations. in Integration of Distributed Energy Resources in Power Systems vol. 2012 51–76 (Elsevier, 2016).
Cropper, M. A. J., Geiger, S. & Jollie, D. M. Fuel cells: A survey of current developments. J. Power Sour. 131, 57–61 (2004).
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Shao, Y., Yin, G. & Gao, Y. Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell. J. Power Sour. 171, 558–566 (2007).
Bing, Y., Liu, H., Zhang, L., Ghosh, D. & Zhang, J. Nanostructured Pt-alloy electrocatalysts for PEM fuel cell oxygen reduction reaction. Chem. Soc. Rev. 39, 2184–2202 (2010).
Platinum, W. & Council, I. Platinum Quarterly Q3 2020. (2020).
Jasinski, R. A new fuel cell cathode catalyst. Nature 201, 1212–1213 (1964).
Zagal, J., Páez, M., Tanaka, A. A., dos Santos, J. R. & Linkous, C. A. Electrocatalytic activity of metal phthalocyanines for oxygen reduction. J. Electroanal. Chem. 339, 13–30 (1992).
Tanaka, A. A., Fierro, C., Scherson, D. & Yaeger, E. B. Electrocatalytic aspects of iron phthalocyanine and its .mu.-oxo derivatives dispersed on high surface area carbon. J. Phys. Chem. 91, 3799–3807 (1987).
Zagal, J. H. & Koper, M. T. M. Reactivity descriptors for the activity of molecular MN4 catalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 55, 14510–14521 (2016).
Zagal, J. H., Griveau, S., Santander-Nelli, M., Granados, S. G. & Bedioui, F. Carbon nanotubes and metalloporphyrins and metallophthalocyanines-based materials for electroanalysis. J. Porphyrins Phthalocyanines 16, 713–740 (2012).
Cao, R. et al. Promotion of oxygen reduction by a bio-inspired tethered iron phthalocyanine carbon nanotube-based catalyst. Nat. Commun. 4, 2076 (2013).
Viera, M. et al. Oxygen reduction reaction at penta-coordinated co phthalocyanines. Front. Chem. 8, 1–12 (2020).
Govan, J. et al. Influence of cyano substituents on the electron density and catalytic activity towards the oxygen reduction reaction for iron phthalocyanine. The case for Fe(II) 2,3,9,10,16,17,23,24-octa(cyano)phthalocyanine. Electrochem. Commun. 118, 106784 (2020).
Ratso, S. et al. High performance catalysts based on Fe/N co-doped carbide-derived carbon and carbon nanotube composites for oxygen reduction reaction in acid media. Int. J. Hydrogen Energy 44, 12636–12648 (2019).
Zagal, J. H., Griveau, S., Silva, J. F., Nyokong, T. & Bedioui, F. Metallophthalocyanine-based molecular materials as catalysts for electrochemical reactions. Coord. Chem. Rev. 254, 2755–2791 (2010).
Randin, J.-P. Interpretation of the relative electrochemical activity of various metal phthalocyanines for the oxygen reduction reaction. Electrochim. Acta 19, 83–85 (1974).
Beck, F. The redox mechanism of the chelate-catalysed oxygen cathode. J. Appl. Electrochem. 7, 239–245 (1977).
Masa, J., Ozoemena, K., Schuhmann, W. & Zagal, J. H. Oxygen reduction reaction using N 4 -metallomacrocyclic catalysts: Fundamentals on rational catalyst design. J. Porphyrins Phthalocyanines 16, 761–784 (2012).
Zhang, R. et al. Oxygen electroreduction on heat-treated multi-walled carbon nanotubes supported iron polyphthalocyanine in acid media. Electrochim. Acta 147, 343–351 (2014).
Zhutaeva, G. V., Bogdanovskaya, V. A., Davydova, E. S., Kazanskii, L. P. & Tarasevich, M. R. Kinetics and mechanism of oxygen electroreduction on Vulcan XC72R carbon black modified by pyrolysis products of cobalt 5,10,15,20-tetrakis(4- methoxyphenyl)porphyrine in a broad pH interval. J. Solid State Electrochem. 18, 1319–1334 (2014).
Zúñiga, C. et al. Elucidating the mechanism of the oxygen reduction reaction for pyrolyzed Fe-N-C catalysts in basic media. Electrochem. Commun. 102, 78–82 (2019).
Zhai, Z. et al. Controlled pyrolysis of ionically self-assembled metalloporphyrins on carbon as cathodic electrocatalysts of polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 46, 11041–11050 (2021).
Li, J. et al. Pyrolysis of self-assembled iron porphyrin on carbon black as core/shell structured electrocatalysts for highly efficient oxygen reduction in both alkaline and acidic medium. Adv. Func. Mater. 27, 1604356 (2017).
Praats, R. et al. Electroreduction of oxygen in alkaline solution on iron phthalocyanine modified carbide-derived carbons. Electrochim. Acta 299, 999–1010 (2019).
Jahnke, H., Schönborn, M. & Zimmermann, G. Organic dyestuffs as catalysts for fuel cells. in Physical and Chemical Applications of Dyestuffs 133–181 (Springer). https://doi.org/10.1007/BFb0046059.
Lefèvre, M., Proietti, E., Jaouen, F. & Dodelet, J.-P. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009).
Jaouen, F. et al. Cross-laboratory experimental study of non-noble-metal electrocatalysts for the oxygen reduction reaction. ACS Appl. Mater. Interfaces. 1, 1623–1639 (2009).
Wu, G. et al. Synthesis–structure–performance correlation for polyaniline–Me–C non-precious metal cathode catalysts for oxygen reduction in fuel cells. J. Mater. Chem. 21, 11392 (2011).
Wu, G., More, K. L., Johnston, C. M. & Zelenay, P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332, 443–447 (2011).
Strickland, K. et al. Highly active oxygen reduction non-platinum group metal electrocatalyst without direct metal-nitrogen coordination. Nat. Commun. 6, 1–8 (2015).
Li, J. et al. Identification of durable and non-durable FeNx sites in Fe–N–C materials for proton exchange membrane fuel cells. Nat. Catal. 4, 10–19 (2021).
Zhao, J. et al. Graphite intercalation compounds derived by green chemistry as oxygen reduction reaction catalysts. ACS Appl. Mater. Interfaces. 12, 42678–42685 (2020).
He, Y., Tan, Q., Lu, L., Sokolowski, J. & Wu, G. Metal-nitrogen-carbon catalysts for oxygen reduction in pem fuel cells: Self-template synthesis approach to enhancing catalytic activity and stability. Electrochem. Energy Rev. 2, 231–251 (2019).
Wei, X. et al. Recent advances in synergistically enhanced single-atomic site catalysts for boosted oxygen reduction reaction. Nano Energy 84, 105817 (2021).
Tylus, U. et al. Elucidating oxygen reduction active sites in pyrolyzed metal-nitrogen coordinated non-precious-metal electrocatalyst systems. J. Phys. Chem. C 118, 8999–9008 (2014).
Jia, Q. et al. Spectroscopic insights into the nature of active sites in iron–nitrogen–carbon electrocatalysts for oxygen reduction in acid. Nano Energy 29, 65–82 (2016).
Jia, Q., Liu, E., Jiao, L., Pann, S. & Mukerjee, S. X-ray absorption spectroscopy characterizations on PGM-free electrocatalysts: Justification, advantages, and limitations. Adv. Mater. 31, 2–9 (2019).
Kramm, U. I. et al. Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 14, 11673–11688 (2012).
Kramm, U. I., Ni, L. & Wagner, S. 57 Fe mössbauer spectroscopy characterization of electrocatalysts. Adv. Mater. 31, 1805623 (2019).
Artyushkova, K., Serov, A., Rojas-Carbonell, S. & Atanassov, P. Chemistry of multitudinous active sites for oxygen reduction reaction in transition metal-nitrogen-carbon electrocatalysts. J. Phys. Chem. C 119, 25917–25928 (2015).
Orellana, W. Catalytic activity toward oxygen reduction of transition metal porphyrins covalently linked to single-walled carbon nanotubes: A density functional study. Phys. Rev. B 84, 155405 (2011).
Orellana, W. Metal-phthalocyanine functionalized carbon nanotubes as catalyst for the oxygen reduction reaction: A theoretical study. Chem. Phys. Lett. 541, 81–84 (2012).
Orellana, W. Catalytic properties of transition metal-N4 moieties in graphene for the oxygen reduction reaction: Evidence of spin-dependent mechanisms. J. Phys. Chem. C 117, 9812–9818 (2013).
Riquelme, J. et al. Biomimicking vitamin B12. A Co phthalocyanine pyridine axial ligand coordinated catalyst for the oxygen reduction reaction. Electrochim. Acta 265, 547–555 (2018).
Luo, F. et al. P-block single-metal-site tin/nitrogen-doped carbon fuel cell cathode catalyst for oxygen reduction reaction. Nat. Mater. 19, 1215–1223 (2020).
Matanovic, I., Artyushkova, K. & Atanassov, P. Understanding PGM-free catalysts by linking density functional theory calculations and structural analysis: Perspectives and challenges. Curr. Opin. Electrochem. 9, 137–144 (2018).
Chung, H. T. et al. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 484, 479–484 (2017).
Niwa, H. et al. Probing carbon edge exposure of iron phthalocyanine-based oxygen reduction catalysts by soft X-ray absorption spectroscopy. J. Power Sources 223, 30–35 (2013).
Kruusenberg, I., Matisen, L. & Tammeveski, K. Oxygen electroreduction on multi-walled carbon nanotube supported metal phthalocyanines and porphyrins in acid media. Int. J. Electrochem. Sci. 8, 1057–1066 (2013).
Ladouceur, M. et al. Pyrolyzed cobalt phthalocyanine as electrocatalyst for oxygen reduction. J. Electrochem. Soc. 140, 1974–1981 (1993).
Kramm, U. I. et al. Effect of iron-carbide formation on the number of active sites in Fe-N-C catalysts for the oxygen reduction reaction in acidic media. J. Mater. Chem. A 2, 2663–2670 (2014).
Visscher, W. & Van Veen, J. A. R. On active-site heterogeneity in pyrolyzed carbon-supported iron porphyrin catalysts for the electrochemical reduction of oxygen: An in situ mo 1 ssbauer study. J. Phys. Chem. https://doi.org/10.1021/jp0266087 (2002).
Han, H. et al. Pyrolysis-driven synthesis of nanoscale carambola-like carbon decorated with atomically dispersed Fe sites toward efficient oxygen reduction reaction. Catal. Sci. Technol. 10, 7160–7164 (2020).
Yamamoto, M. et al. Iron porphyrin-derived ordered carbonaceous frameworks. Catal. Today 364, 164–171 (2021).
Matanovic, I. et al. Core level shifts of hydrogenated pyridinic and pyrrolic nitrogen in the nitrogen-containing graphene-based electrocatalysts: In-plane vs edge defects. J. Phys. Chem. C 120, 29225–29232 (2016).
Holby, E. F. & Zelenay, P. Linking structure to function: The search for active sites in non-platinum group metal oxygen reduction reaction catalysts. Nano Energy 29, 54–64 (2016).
Kneebone, J. L. et al. A combined probe-molecule, Mössbauer, nuclear resonance vibrational spectroscopy, and density functional theory approach for evaluation of potential iron active sites in an oxygen reduction reaction catalyst. J. Phys. Chem. C 121, 16283–16290 (2017).
Glibin, V. P. et al. Non-PGM electrocatalysts for PEM fuel cells: Thermodynamic stability and DFT evaluation of fluorinated FeN 4 -based ORR catalysts. J. Electrochem. Soc. 166, F3277–F3286 (2019).
Kauffman, D. R., Kuzmych, O. & Star, A. Interactions between single-walled carbon nanotubes and tetraphenyl metalloporphyrins: Correlation between spectroscopic and FET measurements. J. Phys. Chem. C 111, 3539–3543 (2007).
Venegas, R. et al. Comparison of the catalytic activity for O2 reduction of Fe and Co MN4 adsorbed on graphite electrodes and on carbon nanotubes. Phys. Chem. Chem. Phys. 19, 20441–20450 (2017).
Abarca, G. et al. In search of the most active MN4 catalyst for the oxygen reduction reaction The case of perfluorinated Fe phthalocyanine. J. Mater. Chem. A 7, 24776–24783 (2019).
Raggio, M. et al. Metallo-corroles supported on carbon nanostructures as oxygen reduction electrocatalysts in neutral media. Eur. J. Inorg. Chem. 2019, 4760–4765 (2019).
Zhang, Y. et al. Cobalt tetramethoxyphenylporphyrin functionalized graphene for oxygen reduction reaction in neutral media. Mater. Lett. 144, 5–8 (2015).
Takase, S., Aoto, Y., Ikeda, D., Wakita, H. & Shimizu, Y. Effects of crystallographic structures of metal-phthalocyanine on electrocatalytic properties of oxygen reduction in acidic condition. Electrocatalysis 10, 653–662 (2019).
Hunter, C. A. & Sanders, J. K. M. The nature of π-π interactions. J. Am. Chem. Soc. 112, 5525–5534 (1990).
Drain, C. M., Varotto, A. & Radivojevic, I. Self-organized porphyrinic materials. Chem. Rev. 109, 1630–1658 (2009).
Kumar, P., Singh, G., Tripathi, D. & Jain, S. L. Visible light driven photocatalytic oxidation of thiols to disulfides using iron phthalocyanine immobilized on graphene oxide as a catalyst under alkali free conditions. RSC Adv. 4, 50331–50337 (2014).
Kuila, T., Mishra, A. K., Khanra, P., Kim, N. H. & Lee, J. H. Recent advances in the efficient reduction of graphene oxide and its application as energy storage electrode materials. Nanoscale 5, 52–71 (2013).
Zhang, Y. et al. Carbon nanotube-zinc oxide electrode and gel polymer electrolyte for electrochemical supercapacitors. J. Alloy. Compd. 480, 17–19 (2009).
Venegas, R. et al. Experimental reactivity descriptors of M-N-C catalysts for the oxygen reduction reaction. Electrochim. Acta 19, 135340 (2019).
Jaouen, F., Lefèvre, M., Dodelet, J. P. & Cai, M. Heat-treated Fe/N/C catalysts for O2 electroreduction: Are active sites hosted in micropores?. J. Phys. Chem. B 110, 5553–5558 (2006).
Kramm, U. I. et al. On an easy way to prepare metal-nitrogen doped carbon with exclusive presence of MeN4-type sites active for the ORR. J. Am. Chem. Soc. 138, 635–640 (2016).
Kumar, Y. et al. Bifunctional oxygen electrocatalysis on mixed metal phthalocyanine- modified carbon nanotubes. ACS Appl. Mater. Interfaces https://doi.org/10.1021/acsami.1c06737 (2021).
Prieto, P., De La Figuera, J., Sanz, J. M. & Marco, J. F. Effects of low energy ion bombardment on the formation of cubic iron mononitride thin films. Thin Solid Films 539, 35–40 (2013).
Briggs, D. & Gibson, V. A. Direct observation of multiplet splitting in 2P photoelectron peaks of cobalt complexes. Chem. Phys. Lett. 25, 493–496 (1974).
Frost, D. C., McDowell, C. A. & Woolsey, I. S. X-ray photoelectron spectra of cobalt compounds. Mol. Phys. 27, 1473–1489 (1974).
Bard, A. J. & Faulkner, L. R. Electrochemical Method. Fundamentals and Applications (Wiley, 2001).
Tammeveski, K. & Zagal, J. H. Electrocatalytic oxygen reduction on transition metal macrocyclic complexes for anion exchange membrane fuel cell application. Curr. Opin. Electrochem. 9, 207–213 (2018).
Thorum, M. S., Hankett, J. M. & Gewirth, A. A. Poisoning the oxygen reduction reaction on carbon-supported Fe and Cu electrocatalysts: Evidence for metal-centered activity. J. Phys. Chem. Lett. 2, 295–298 (2011).
Zagal, J. H., Bedioui, F. & Dodelet, J. P. N4-Macrocyclic Metal Complexes (Springer, 2006). https://doi.org/10.1007/978-0-387-28430-9.
Zagal, J. H., Gulppi, M., Isaacs, M., Cárdenas-Jirón, G. & Aguirre, M. J. Linear versus volcano correlations between electrocatalytic activity and redox and electronic properties of metallophthalocyanines. Electrochim. Acta 44, 1349–1357 (1998).
Cárdenas-Jirón, G. I. et al. Reactivity of electrodes modified with substituted metallophthalocyanines. Correlations with redox potentials, Hammett parameters and donor-acceptor intermolecular hardness. Electrochim. Acta 46, 3227–3235 (2001).
Ramaswamy, N. & Mukerjee, S. Influence of inner- and outer-sphere electron transfer mechanisms during electrocatalysis of oxygen reduction in alkaline media. J. Phys. Chem. C 115, 18015–18026 (2011).
Zúñiga, C. et al. Reactivity indexes for the electrocatalytic oxidation of hydrogen peroxide promoted by several ligand-substituted and unsubstituted Co phthalocyanines adsorbed on graphite. J. Electroanal. Chem. 765, 22–29 (2016).
Choi, C. H. et al. The Achilles’ heel of iron-based catalysts during oxygen reduction in an acidic medium. Energy Environ. Sci. 11, 3176–3182 (2018).
Ramaswamy, N., Tylus, U., Jia, Q. & Mukerjee, S. Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: Linking surface science to coordination chemistry. J. Am. Chem. Soc. 135, 15443–15449 (2013).
Jia, Y., Jiang, K., Wang, H. & Yao, X. The role of defect sites in nanomaterials for electrocatalytic energy conversion. Chem 5, 1371–1397 (2019).
Zhang, J. et al. Defect and doping co-engineered non-metal nanocarbon ORR electrocatalyst. Nano-Micro Lett. 13, 65 (2021).
Pegis, M. L., Wise, C. F., Martin, D. J. & Mayer, J. M. Oxygen reduction by homogeneous molecular catalysts and electrocatalysts. Chem. Rev. 118, 2340–2391 (2018).
Silva, N. et al. Probing the Fen+/Fe(n−1)+ redox potential of Fe phthalocyanines and Fe porphyrins as a reactivity descriptor in the electrochemical oxidation of cysteamine. J. Electroanal. Chem. 819, 502–510 (2018).
Fletcher, S. Tafel slopes from first principles. Solid State Electrochem 13, 537–549 (2009).
Huang, X. et al. Efficient oxygen reduction catalysts of porous carbon nanostructures decorated with transition metal species. Adv. Energy Mater. 10, 1–21 (2020).
Zhang, J., Yang, H. & Liu, B. Coordination engineering of single-atom catalysts for the oxygen reduction reaction: A review. Adv. Energy Mater. 11, 1–20 (2021).
Wang, X. et al. Review of metal catalysts for oxygen reduction reaction: From nanoscale engineering to atomic design. Chem 5, 1486–1511 (2019).
Kruusenberg, I. et al. Cobalt-nitrogen co-doped carbon nanotube cathode catalyst for alkaline membrane fuel cells. ChemElectroChem 3, 1455–1465 (2016).
Govan, J., Orellana, W., Zagal, J. H. & Tasca, F. Penta-coordinated transition metal macrocycles as electrocatalysts for the oxygen reduction reaction. J. Solid State Electrochem. 25, 15–31 (2021).
Zagal, H. et al. A possible interpretation for the high catalytic activity of heat-treated non-precious metal Nx / C catalysts for O 2 reduction in terms of their formal potentials. Electrochem. Solid-State Lett. 15, B90–B92 (2012).
Zagal, J. H. et al. Towards a unified way of comparing the electrocatalytic activity MN4 macrocyclic metal catalysts for O2 reduction on the basis of the reversible potential of the reaction. Electrochem. Commun. 41, 24–26 (2014).
Giannozzi, P. et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Langreth, D. C. et al. A density functional for sparse matter. J. Phys.: Condens. Matter 21, 084203 (2009).
Thonhauser, T. et al. Van der Waals density functional: Self-consistent potential and the nature of the van der Waals bond. Phys. Rev. B 76, 125112 (2007).
Román-Pérez, G. & Soler, J. M. Efficient implementation of a van der waals density functional: Application to double-wall carbon nanotubes. Phys. Rev. Lett. 103, 1–4 (2009).
Thonhauser, T. et al. Spin signature of nonlocal correlation binding in metal-organic frameworks. Phys. Rev. Lett. 115, 1–6 (2015).
Berland, K. et al. van der Waals forces in density functional theory: A review of the vdW-DF method. Rep. Prog. Phys. 78, 066501 (2015).
Garrity, K. F., Bennett, J. W., Rabe, K. M. & Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 81, 446–452 (2014).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Ma, G. et al. Nitrogen-doped hollow carbon nanoparticles with excellent oxygen reduction performances and their electrocatalytic kinetics. J. Phys. Chem. C 115, 25148–25154 (2011).
Paulus, U. A., Schmidt, T. J., Gasteiger, H. A. & Behm, R. J. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disk electrode study. J. Electroanal. Chem. 495, 134–145 (2001).
Acknowledgements
We thank financial support from FONDECYT Projects No. 1181840, 1220988 1170480 Dicyt-USACH publicaciones and DICYT POSTDOC 20211768. The computational resources used in this work were provided by Fenix HPC (UNAB).
Author information
Authors and Affiliations
Contributions
W.O. is responsible for the theoretical conceptualization and calculations, data interpretation and the corresponding writing. C.Z., is responsible for the synthesis of the complexes and all the experimental part and the corresponding writing. J.M. is responsible for all the spectroscopic experiments, analysis and the corresponding writing. F.T. is responsible for the conceptualization of the whole project, supervision of the experimental part, final writing revision and submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Orellana, W., Loyola, C.Z., Marco, J.F. et al. Evidence of carbon-supported porphyrins pyrolyzed for the oxygen reduction reaction keeping integrity. Sci Rep 12, 8072 (2022). https://doi.org/10.1038/s41598-022-11820-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11820-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
