
Abstract
Sorbus pohuashanensis (Hance) Hedl. is a Chinese native alpine tree species, but the problem of introducing S. pohuashanensis to low altitude areas has not been solved. In this study, we aimed to explore the molecular regulatory network of S. pohuashanensis in response to high-temperature stress using RNA-Sequencing technology and physiological and biochemical determination. Based on transcriptomic data, we obtained 1221 genes (752 up-regulated and 469 down-regulated) that were differentially expressed during 8 h 43℃ treatment and candidate genes were related to calcium signaling pathway, plant hormone signal transduction, heat shock factors, chaperones, ubiquitin mediated proteolysis, cell wall modification, ROS scavenging enzymes, detoxification and energy metabolism. The analysis of high temperature response at the physiological level and biochemical level were performed. The chlorophyll fluorescence parameters of leaf cells decreased, the content of osmotic regulators increased, and the activity of ROS scavenging enzymes decreased. The molecular regulatory network of S. pohuashanensis in response to high-temperature stress was preliminarily revealed in this study, which provides fundamental information improving introducing methods and discovering heat-tolerant genes involved in high-temperature stress in this species and provides a reference for other plants of the genus Sorbus.
Similar content being viewed by others
Introduction
Introduction and acclimatization is the nearest way to enrich species and breeding materials, which is widely used worldwide1. The introduction and acclimatization of alpine plants has always been one of the difficulties in forest genetics2. Wild species of alpine plants mainly grow in the mountains at an altitude of several thousand meters3. Due to the mountain forest environment, alpine plants have special requirements for light, temperature, water, soil and other conditions, and temperature, especially high-temperature, is one of the most important factors that restrict the success of introduction and acclimatization4. Heat stress can be classified into moderate and extreme according to duration of stress, time of the day at which it occurs and co-exposure to other stresses, each of which involves different coping mechanisms and adaptation strategies5. Such temperature stress occurs regularly in alpine environments6. Recently, increasing numbers of reports have indicated that with climate warming, the high temperature heat wave will last longer and will occur more frequently7. In addition, plants are more often exposed to severe high-temperature stress because they are fixed and high-temperature affects the physiological and ecological processes of plants8. Therefore, studying and discussing the thermal tolerance of plants and discussing how to improve the thermal tolerance of plants has become one of the important topics to cope with global warming, and has gradually received widespread attention. Previous reports have shown that attempts to introduce alpine plants have made some advance. For example, the tolerance of Trollius chinensis to extreme high temperatures in summer can be improved by shading seedlings after being introduced to the Beijing plain from the alpine mountainous area of 1500–2600 m above sea level9. The high temperature and drought in the introduced area greatly affect the growth and development of Rhododendron lapponicum. However, the stress resistance of R. lapponicum can be increased by breeding varieties with better heat tolerance10. But the specific mechanisms that influence the heat resistance of alpine plants after introduction to the plains are still unclear.
Existing studies show that adverse effects of high-temperature stress on plants mainly include inhibition of seed germination, reduction of plant growth and reproduction, and reduction of crop yield and quality11. High temperature stress affects all aspects of plant life, with photosynthesis taking the brunt of the stress and being highly susceptible to heat-mediated damage.Though several chloroplast proteins get denatured or become nonfunctional at high-temperature, the photosystem-II (PSII) is most vulnerable to get damaged12. High-temperature damages the chloroplast and mitochondrial electron transport system. It generates ROS such as superoxides, hydrogen peroxide (H2O2), and hydroxyl radicals (·OH) that harm DNA and also cause lipid peroxidation of the cell membrane13. Plant response to high-temperature stress is relatively conservative14. Previous studies have revealed some key factors in the plant response system to high-temperature stress, which together constitute the main regulatory network of plant response to high-temperature stress. Plant response to high-temperature stress regulation network including small G protein/G protein mediated calcium signaling pathways, hormone regulation networks, heat shock transcription factors (Hsfs) and heat shock proteins (Hsps) reaction and reactive oxygen species (ROS) reaction, and these factors interact together to complete the response of high-temperature stress15. Plant cells first perceive high-temperature stimulation and amplify intracellular signals, then induce the expression of heat-resistant genes. Signal transduction and transcription regulation networks are crucial in the process of plant responses to high-temperature stress16. Calcium and calmodulin (CaM) are locatedupstream of the high temperature signal transmission chain and have the characteristics of rapidity and broad spectrum. High-temperature triggered signals promote calcium to bind to CaM, further activate calcium-dependent protein kinase (CDPK), and induce mitogen-activated protein kinases (MAPKs) transmit signals to the nucleus and ultimately induce heat-tolerant gene expression17. Heat shock transcription factor A1s (HsfA1s) is considered to be a key "regulator" of thermotolerance, which can activate the expression of heat shock response (HSR) genes, and then regulate the synthesis of molecular chaperones and enzymes involved in unfolded protein degradation and ROS scavenging18. Hsps play the most crucial role in the several intricate mechanisms which plants protect themselves from excessive high-temperature stress19,20. High temperatures lead to the accumulation of unfolded proteins in plant cells, which must be regenerated or degraded by plants in order to maintain normal life activities21. By functioning as molecular chaperones, Hsps prevent protein denaturation and aggregation. In addition, Hsps interact and modulates functions of heat shock factors (HSFs) e.g. HsfAs, DREB, bZIP and WRKY22. ROS primarily functions as signal transduction molecules that regulate different pathways during plant acclimation to stress, but are also toxic byproducts of stress metabolism13. Accumulation of ROS in plants activates HSFs, which in turn activate ROS scavenging and detoxifying enzymes like ascorbate peroxidase (APX) and superoxide dismutase (SOD)13. Catalase (CAT) are also considered to be ROS scavenging enzymes necessary for ROS detoxification23. A decrease in ROS level occurs due to the enhanced production of antioxidants, osmolytes, and Hsps. The major stress-responsive osmolytes in plants include proline (Pro), glycine betaine, and trehalose, which play roles in maintaining cellular ionic homeostasis24. In addition, plant tissues also generate adaptive structures to resist the adverse effects of high-temperature. As a powerful dynamic barrier, cell wall can make metabolic and structural adaptations to the physiological changes of cells caused by high temperature stress, including the formation of cell wall modifications such as the cuticle in the aerial part and suberin in the root, and produce a special phenol ester-based protection system25.
Sorbus pohuashanensis (Hance) Hedl. , a small deciduous tree of the subfamily Maloideae of Rosaceae, is a native tree species and widely distributed at altitudes of 900–2,500 m in northern China26. With beautiful flowers, colorful fruits, and leaves of different colors in the four seasons, S. pohuashanensis has a very high ornamental significance in landscape27. In addition, S. pohuashanensis has specific medicinal and commercial significance26,28. Research on S. pohuashanensis has in recent years focused on plant secondary structure and affinities29, phylogenetic relationships of pink-fruit30, seed germination and dormancy characteristics31, nutritional and medicinal components of the fruit26,32, identification of pathogenic pathogens and development and application of SSR markers27,33. However, the main problem facing the development and utilization of S. pohuashanensis is the poor adaptability of introduction and acclimatization to the high temperature environment in summer in low altitude areas. Peng et al. analyzed the heat resistance physiological characteristics of S. pohuashanensis and found that it was sensitive to high-temperature stress34. At the molecular level, the mechanisms of high temperature response in S. pohuashanensis have been initially explored. Liu et al. sequenced the transcriptome of S. pohuashanensis and obtained a large number of Unigenes, from which SpHsp70-1 was cloned to investigate the mechanisms and expression patterns in response to high temperature27,35. Zhang et al. cloned the small heat stress proteins(sHsps) SpHsp17.3 and SpHsp23.8 from S. pohuashanensis and investigated their tissue-specific expression and expression in response to abiotic stresses36,37. However, the molecular regulatory mechanisms of the response of S. pohuashanensis to high-temperature stress have not been elucidated. In this study, the differentially expressed genes (DEGs) of S. pohuashanensis were analyzed and the molecular regulatory networks were constructed during high-temperature treatment using RNA-seq technology to reveal the molecular response mechanisms to high temperature with determination of chloroplast fluorescence parameters, osmotic regulation substance content, and ROS scavenging enzyme activity. The results will contribute to the development and utilization of germplasm resources for S. pohuashanensis and provide an informatics basis for the genetic improvement and breeding of varieties of this species in the future.
Results
Phenotypes of S. pohuashanensis leaves under normal and high-temperature stress
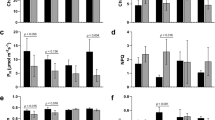
The PSII process of dark reaction of photosynthesis in leaves of S. pohuashanensis in HT was affected to some extent within 0–8 h under 43℃ (Fig. 1). Fv/Fm of leaves in HT was lower than that in CK from 0 h onwards, and Fv/Fm of leaves in CK remained above 0.84 (Fig. 1a). The Fv '/Fm'of the leaves in the HT showed a decreasing trend and was significantly lower than that in the CK, while the Fv '/Fm' in the CK showed a rising and then decreasing trend (Fig. 1b). In addition, PSII of S. pohuashanensis leaves in HT showed a downward trend with the extension of time under high temperature stress, which was significantly lower than CK after 4 h of high temperature stress (Fig. 1c). There was no difference in qL between HT and CK in the first 6 h of high temperature stress, and HT was significantly lower than CK at 6–8 h of high temperature stress (Fig. 1d). It is worth mentioning that the NPQ and qN of the leaves in HT were always lower than those in CK after the beginning of high temperature stress, and gradually decreased with the extension of time; while the NPQ and qN in CK changed little within 0–8 h, and basically remained above 0.860 and 0.520 (Fig. 1e, f). The above results indicated that PSII process of HT was inhibited compared with CK.

Effects of heat stress on the (a) Fv/Fm, (b) Fv`/Fm`, (c) ΦPSII, (d) qL, (e) NPQ and (f) qN of PS IIphotochemistry (significant difference at 5% levels according to standard deviation and multiple comparison).
High temperature stress affected the content of osmotic regulatory substances in the leaves of S. pohuashanensis. After 8 h of high temperature stress, the content of malondialdehyde (MDA) in HT increased sharply, by 60.13% compared with CK (Fig. 2a) indicating the membrane lipid showed much higher degree of damage in HT. The Pro content in the leaves of S. pohuashanensis in HT also increased sharply, which was significantly different from the CK and 143.90% higher than that in CK (Fig. 2b), and the soluble sugar content and soluble protein content in HT were 17.79% and 5.75% higher than CK, respectively (Fig. 2c, d). Compared with CK, the content of osmoregulation substances in HT leaves increased.

Physiological and biochemical indexes of leaves of S. pahuashanensis under heat stress. (a) MDA, (b) Pro, (c) Soluble sugar, (d) Total soluble protein, (e) CAT, (f) POD, (g) SOD and (h) APX (* indicate the significant difference at 5% levels according to student’s t-test, respectively. NS means no significant difference).
High temperature stress also significantly affected the activity of ROS scavenging enzymes. Except for CAT, POD, SOD and APX activities of the leaf cells of S. pohuashanensis were significantly lower in HT than in CK after 8 h of high temperature stress (Fig. 2e, f, g, h).
Transcriptome sequencing and de novo assembly
To elucidate the molecular responses to heat stress in S. pohuashanensis, six libraries were constructed from HT and CK samples for sequencing using the Illumina HiSeq2000 platform. In this study, approximately 53,461,517 raw reads and 51,558,437 clean reads were generated per sample (Supplementary Table S1). A total of 229,107 contigs were obtained after preliminary assembly. These contigs were assembled using the scaffolding algorithm (Word size = 45, Minimum contig length ≥ 200) of CLC Genomics Workbench software, and 197,028 primary unigenes were obtained. Finally, using CAP3 online stitching software, the primary unigenes were sequenced a second time and a transcriptome database for S. pohuashanensis was obtained. The de novo assembly generated 130,003 final unigenes, with an average length of 658 bp and an N50 of 838 bp (Supplementary Table S2). The completeness of the transcriptome assembly was assessed using BUSCO, which is based on evolutionarily informed expectations of gene content from eukaryota_odb10. Compared to the 255 single-copy orthologs for the embryophyta lineage, our assembly was 49.1% complete (107 complete single-copy and 18 complete duplicated BUSCO), while 37.3% of contigs were fragmented (95 BUSCOs) and 13.6% were missing (35 BUSCOs). These results indicated that the transcriptome assembly was useful for further transcriptomic analyses of S. pohuashanensis (Supplementary Fig. S1).
GO and KEGG pathway analysis of DEGs
FPKM was used to calculate the gene expression levels among different samples and to compare the mRNA levels of S. pohuashanensis response to high-temperature stress (Supplementary Table S1). DEGs (fold change ≥ 2 and q-value ≤ 0.05) were defined as genes that were abundant or rare in one sample relative to the other sample. The results showed that 1221 DEGs were identified in S. pohuashanensis after high-temperature stress (752 upregulated and 469 downregulated) (Fig. 3; Supplementary Table S4).

Volcano plot of differentially expressed transcripts with high-temperature stress treatment in S. pohuashanensis at q-value ≤ 0.05. Up-regulated and down-regulated genes were represented by red and blue dots, respectively.
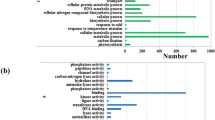
Comparing the 130,003 final unigenes with the Nr database, a total of 71,633 unigenes were annotated, accounting for approximately 55.1% of the total unigenes (Supplementary Table S5). To identify the DEGs significantly enriched for GO terms, the function of DEGs after high-temperature stress was analyzed using Web-based agriGO software. Among the 1,221 differentially expressed genes, 827 DEGs were significantly enriched for 56 GO terms (q-value ≤ 5%) (Supplementary Fig. S2; Table S6). In the biological process category, metabolic processes (GO: 0,008,152) (n = 72) and oxidation–reduction process (GO: 0,055,114) (n = 49) were the most abundant subcategories. In the cell components category, extracellular regions (GO: 0,005,576) (n = 8) and mitochondrion (GO: 0,005,739) (n = 6) were the most abundant subcategories. In the molecular function category, metal ion binding (GO: 0,046,872) (n = 77), zinc ion binding (GO: 0,008,270) (n = 77), and nucleic acid binding (GO: 0,003,676) (n = 69) were abundant subcategories (Fig. 4; Supplementary Table S6).

GO enrichment analysis of DEGs under high-temperature treatment. The figure shows the top 30 terms, and the size of the points indicates the number of differentially expressed genes involved in the path. The color scale indicates the significance level (q-value ≤ 0.05). The rich factor is the ratio between the number of DEGs and all genes enriched for the terms.
To further explore DEGs involved in the biological pathway and signal transduction pathway of S. pohuashanensis, the number of DEGs in each KEGG pathway was estimated. A total of 1,221 DEGs were assigned to the 151 KEGG pathways (q-value ≤ 0.05). These pathways were mainly related to the carbohydrate metabolism, biosynthesis of other secondary metabolites, lipid metabolism, energy metabolism, amino acid metabolism, transport and catabolism, endocrine system, signal transduction, metabolism of terpenoids and polyketides, folding, sorting and degradation, and metabolism of cofactors and vitamins (Fig. 5; Supplementary Table S7). According to the FPKM value of DEGs and the enrichment annotation of DEGs in go and KEGG, DEGs were screened and further analyzed according to signal transduction, transcriptional regulation, ROS homeostasis and protein homeostasis, and metabolic process.

KEGG enrichment analysis of differentially expressed genes under high-temperature treatment. The figure shows the top 30 pathways. The size of the dot indicates the number of DEGs involved in the pathway. The color scale indicates the significance level (q-value ≤ 0.05). The rich factor is the ratio between the number of DEGs and all genes enriched for the pathways.
Differential expression analysis of genes related to signal transduction
Among the 1,221 DEGs, 41 putative genes correlated with signal transduction for high-temperature stress were identified (Supplementary Table S8). In this study, the signal transduction pathways of plant response to high-temperature stress include calcium signaling transduction, RLKs, phosphatidylinositol signaling, mitogen activated protein kinase (MAPK) signaling, cyclic adenosine monophosphate (cAMP) signaling, phospholipase D (PLD) signaling and plant hormones.
Calcium signaling
After further analysis, 11 DEGs were found to be involved in the calcium signaling transduction pathway. Calcium signaling-related genes CBL10, CCAMK-like and MCUb were up-regulated, and CPK26-like, CML50, PBP1-like and G protein-related genes ARF, BIG5-like, FTSZ1-like, NOA1, and SAR1A were down-regulated under high temperature stress (Fig. 6; Supplementary Table S8).

taken from high-temperature treatment (HT) and control (CK). The changes in expression are indicated by colors ranging from blue (down-regulated) to yellow red (up-regulated).
Relative expression levels of DEGs related to signal transduction pathway. The rows in the heat map represent the individual gene IDs, and the columns represent the samples
Receptor-like kinase
Eleven DEGs belong to receptor-like kinase (RLK) were found, in particular, leucine-rich repeat receptor-like kinase (LRR-RLK) genes. In the LRR-RLK, there were 2 genes upregulated (NIK1 and LOC103432629-like) and 9 (CEPR1-like, CLV1-like, LOC114827494-like, LOC103427118-like, LOC103400642-like, Lr10-like, LRR-RLK, RLK7, and RLP12) were downregulated (Fig. 6; Supplementary Table S8).
Intracellular signaling pathways
Moreover, 10 genes related to intracellular signaling pathways were identified. In the phosphatidylinositol signaling pathway, only PLC2-like was upregulated and 6 genes, ARF, BIG5-like, FTSZ1-like, NOA1, PLC2-like, and SAR1A were downregulated. In the MAPK signaling pathway, YDA-like was upregulated and UBC28-like was downregulated. In thecAMP signaling pathway, PLDδ-like was upregulated and ACX1-like was downregulated. In the PLD signaling pathway, only PLD δ-like was up-regulated (Fig. 6; Supplementary Table S8).
Plant hormone
Interestingly, 10 genes involved in plant hormone synthesis have also been identified from DEGs, UVR8-like, a gene related to HY5 regulation, GA2-like associated with gibberellin (GA) precursor synthesis, NCED1-like, and NCED3-like related to abscisic acid (ABA) synthesis were upregulated. However, ABP19A, IAA6, GAI-like, GASA1-like, SAMT-like, PED1, and BSK2-like were downregulated (Fig. 6; Supplementary Table S8).
Differential expression analysis of genes related to transcription regulation
As the most important regulators in plants, HSFs play an important role in the transcriptional regulatory network of plant responses to high-temperature stress. In this study, 26 DEGs were identified related to TFs (Supplementary Table S9).
HSFs
After further analysis, 9 HSFs (e.g., Hsf, Hsp, BIP, bZIP, and WRKY) response to high temperature stress were identified. Two Hsf genes, HsfA3-like NF-YB3, and 2 Hsp genes, Hsp70-like and sHsp-like, were also found to be upregulated. Two down-regulated BIP genes (BIP5 and BIP5-like) were found. However, only one b-ZIP gene (bZIP60) and one WRKY gene (GL2-like) appeared to be DEGs (Fig. 7, Supplementary Table S9).

taken from high-temperature treatment (HT) and control (CK). The changes in expression are indicated by colors ranging from blue (down-regulated) to yellow red (up-regulated).
Relative expression levels of DEGs related to transcriptional regulation. The rows in the heat map represent the individual gene IDs, and the columns represent the samples
TFs
Eleven TFs response to high temperature stress were identified. NAC genes were found to be upregulated (NAC2, NAC29, NAC2-like, NAC29-like, NAC68-like, and Nac72) and one (NAC12-like) was downregulated. One upregulated ATHB gene (ATHB12-like), one down-regulated ATHB gene (ATHB6-like), one upregulated ERF gene (ERF011-like), and one down-regulated ERF gene (ERF113) were found. (Fig. 7, Supplementary Table S9).
F-box
Six genes of F-box protein, a key component of the E3 ubiquitin ligase SCF complex, were identified in DEGs. Three upregulated F-box/kelch repeat protein genes (LOC103964362-like, LOC103409567-like, and LOC103451255-like), 2 upregulated F-box/LRR-repeat protein genes (FBL14-like and MAX2A), and F-box protein gene (SKIP23-like) were identified (Fig. 7, Supplementary Table S9).
Differential expression analysis of genes related to protein homeostasis and ROS homeostasis
In this study, 70 putative genes correlated with protein homeostasis and ROS homeostasis for high-temperature stress were identified (Fig. 8, Supplementary Table S10), which mainly involved cell wall, early secretary pathway, active transport of plasma membrane, chaperone, autophagy, detoxification, enzymatic antioxidant system.


taken from high-temperature treatment (HT) and control (CK). The changes in expression are indicated by colors ranging from blue (down-regulated) to yellow red (up-regulated).
Relative expression levels of DEGs related to protein homeostasis (a) and ROS homeostasis (b). The rows in the heat map represent the individual gene IDs, and the columns represent the samples
Cell wall
Fourteen DEGs were found to be involved in the cell wall synthesis and modification and 5 were up-regulated, GAUT3, GRPA3-like and HHT1-like, while 9 were down-regulated CER26-like, GRP-like, PME35, PME-like, TBL1, TBL6, TBL19, TBL38 and XTH23(Fig. 8, Supplementary Table S10).
Plasma membrane and early secretary pathway
In the plasma membrane and transmembrane transport, 18 DEGs were identified. Glutamate receptor protein (GLR) gene GLR2.2 and GLR2.8 were up-regulated. Dynein genes XI-E-like and VIII-1 were up-regulated; aquaporin genes TIP1-3 and TIP1-3-like, transporter genes abcG11-like, abcG22-like, TT12, TT12-like, NPF3.1, NPF3.1-like, NPF5.1and NPF5.6, coatomer subunit gene COPB1-like, ER-GA-transported protein complex gene SAR1A, vacuolar transport-related genes GSNAP and VSR6-like, endocytosis-related gene EHD1-like, dynein gene VIII-E-like, galacturonidase-related gene LUG-like were down-regulated (Fig. 8, Supplementary Table S10).
Chaperone and autophagy
In addition to protein homeostasis-related genes, 12 DEGs were identified, chaperone DnaJ genes ATJ11-like and ATJ6-like, Bcl-2-associated athano gene BAG2-like, disease resistance protein genes RGA2-like and RGA3 and vicilin gene AMP2-2 were up-regulated and disease resistance protein gene RGA2-like, autophagy gene ATG8C and universal stress protein gene uspA-like were down-regulated (Fig. 8, Supplementary Table S10).
CYP450
DEGs related to ROS homeostasis were also identified. In the detoxification, 7 genes, CYP71A1-like, CYP71AV8-like, CYP749A22-like, CYP77A1-like, CYP81D11-like, AKR4C9-like and idnD-like were up-regulated and 11 genes, CYP714C2-like, CYP71A1-like, CYP72A219-like, CYP79A1-like, CYP79D3, CYP82A3-like, CYP82G1-like, CYP85A, CYP90A1, CYP90A1-like and CYP94C1-like were down-regulated (Fig. 8, Supplementary Table 10).
ROS scavenger enzyme
In the ROS scavenger enzyme-related genes, only GSTU9-like was up-regulated and 7 genes, GSTU17-like, MDAR5, PER47, PER47-like, PERP7-like and PNC1-like were down-regulated (Fig. 8, Supplementary Table S10).
Differential expression analysis of genes related to metabolism process
In this study, 58 genes related to material and energy metabolic processes in DEGs were identified. mainly involving carbohydrate metabolism, lipid metabolism, amino acid metabolism, photosynthesis, respiration (Fig. 9, Supplementary Table S11).

taken from high-temperature treatment (HT) and control (CK). The changes in expression are indicated by colors ranging from blue (down-regulated) to yellow red (up-regulated).
Relative expression levels of DEGs related to metabolism process. The rows in the heat map represent the individual gene IDs, and the columns represent the samples
Carbohydrate metabolism
Seventeen genes related to carbohydrate metabolism were identified, and 7 genes, BAM1, BAM1-like, BGAL, GAPB, GBSS1-like, UGE1 and UGE1-like were up-regulated, while 10 genes, AMY2, GBSS1-like, GWD2, LSF2, PP2A4-like, PP2A9-like, GML, UGT85A2-like, UGT87A1-like and PDCB4 were down-regulated (Fig. 9, Supplementary Table S11).
Lipid metabolism
In lipid metabolism, 9 genes were identified from DEGs and SDRB-like and GDE1-like were up-regulated, while 7 genes, GDSL-like, PES2-like, CXE15, GPP2, LACS2, LACS4 and PXG4 were down-regulated (Fig. 9, Supplementary Table S11).
Nitrogen metabolism and amino acid metabolism
In nitrogen metabolism and amino acid metabolism, 9 genes were also detected from DEGs, and 5 genes, PYD2-like, ARD1-like, ASN-like, GDH2 and ERVB-like were up-regulated and 4 genes, NIA, NIA-like, CLPP-like and TAT2 were down-regulated (Fig. 9, Supplementary Table S11).
Energy metabolism
Eighteen genes related to energy metabolism were also identified from DEGs. Seven genes related to electron transport chain were found and four respiratory electron transport chain genes, cob, cox1, cox2, cox3 were up-regulated, while only one photosynthetic electron transport chain gene, LFNR-like was down-regulated. In respiration, entire 4 genes, BCS1-A, BCS1-like, FTSH4, MPC2-like were down-regulated. In photosynthesis, only PSBR-like was up-regulated and 2 genes, NADP-ME and TKL were down-regulated (Fig. 9, Supplementary Table S11).
Quantitative RT-PCR determination of relative DEGs
In order to verify the accuracy of the transcriptome sequencing results, 20 genes were randomly selected for quantitative RT-PCR verification (Fig. 10a). The results showed that the relative expression patterns of these genes in the HT and CK groups were consistent with those obtained by RNA-Seq (Supplementary Table S4). In addition, we determined their response functions at high temperatures. Comparison of transcriptomic data and qRT-PCR results showed a high correlation (R2 = 0.9345) (Fig. 10b).

Relative gene expression of DEGs analyzed by quantitative RT-PCR in response to high-temperature stress treatment. (a) quantitative RT-PCR data were normalized using the S. pohuashanensis Actin β gene and are shown relative to CK (normal condition). X-axes show different treatments (CK, normal condition and HT, high temperature condition) and Y-axes are scales of relative expression level (error bars indicate SD). (b) Correlation of expression levels between RNA-seq and qRT-PCR that identified 20 HT vs. CK. The log2qRT-PCR (y-axis) was plotted against log2RNA-seq (x-axis).
Discussion
Temperature factor is one of the most important factors which can affect the growth and development of plants and limit the geographical distribution of plants38. It is particularly important to explore the molecular mechanism of plant response to high temperature during introduction and acclimatization. Based on Illumina HiSeq2000 platform, this study conducted RNA-seq on leaves of S. pohuashanensis, and obtained high-quality transcriptome data responding to high-temperature stress. A total of 1,221 DEGs were obtained, among which 752 were up-regulated and 469 were down-regulated. DEGs were mainly concentrated in calcium signal transduction, RLKs, TFs, cell wall and membrane system, chaperone, ROS and metabolism, indicating that high-temperature stress has a multi-aspect effect on life activities of S. pohuashanensis.
The signal transduction pathways of plant response to high-temperature stress include calcium signaling transduction, phosphatidyl inositol signaling, MAPK signaling, cAMP signaling, PLD signaling, RLKs, and plant hormone signal transduction22. High-temperature stress could affected the transport of Ca2+ on the plasma membrane of S. pohuashanensis and promotes the function of G proteins/small G proteins. The increase in intracellular Ca2+ concentration activates the G-protein-mediated phosphatidyl inositol signaling pathway (G-protein-PLC-PKC)39. PLC located on the cell membrane can combine with SA to participate in the response to high-temperature stress, and its activity reaches the maximum after 40 min of high-temperature treatment in Pisum sativum40,41. In this study, PLC2-like was upregulated (Supplementary Table S4; Table S8), indicating that PLC might play a specific role in the response to high-temperature stress in S. pohuashanensis. When ligands (hormones, neurotransmitters, and growth factors, etc.) bind to specific receptors on the cell membrane, adenylate cyclase is activated through G protein-coupled receptors to increase the production of cAMP. Then, cAMP-activated protein kinase A (PKA). PKA regulates the activities of various proteins, including TFs, through phosphorylation, so as to regulate a variety of physiological molecules in cells such as ion channels, cytoskeleton proteins, and enzymes42. PLDδ may regulate signal transduction in the process of plant stress resistance through phosphatidic acid (PA), an important signaling molecule in plant cell signal transduction43. PLD can also directly interact with heterotrimer G protein to influence cell signal transduction44. Moreover, we found that genes related to intercellular and intracellular signaling pathways, MAPK signaling pathways (YDA-like), cAMP signaling pathways, and PLD signaling pathways (PLDδ) were up-regulated (Supplementary Table S4, Table S9), which implies that S. pohuashanensis could transports high-temperature signals to the nucleus through the MAPK, cAMP, and PLD pathways, further activating hormone signaling and TF regulation.
In our study, HSFs (HsfA3-like, BIP5, BIP5-like, bZIP60, GL2-like, sHsp-like, Hsp70-like and Hsp70-17-like) and other TF (ATHB6-like, ATHB12-like, ERF011-like, ERF113) genes were found in the high-temperature transcriptome of S. pohuashanensis (Supplementary Table S4; Table S9). High-temperature stress causes damage to membrane proteins, denaturation and inactivation of various enzymes, and accumulation of ROS leading to cell injury and death. Hsfs function as transcriptional activators for several genes including Hsps, regulating the expression of Hsps to maintain homeostasis in plants against heat and chemical stresses45. The expression of Hsps by Hsfs genes is regulated via their interactions with a palindromic binding motif in the promoter region of heat-responsive genes, such as heat shock elements to counteract heat stress-induced ROS46. Class A Hsfs function as central activators of the HSR, and HsfAs can activate the expression of HSR genes and regulate the synthesis of chaperones and enzymes involved in unfolded protein degradation and ROS scavenging47. Under normal and long-term heat stress conditions, they are strictly regulated by various mechanisms to avoid detrimental effects upon activation or over-accumulation18. In a recent report, both LlHSFA3A and LlHSFA3B of Lilium spp. are induced by heat stress. Overexpressing LlHsfA3A in Arabidopsis enhanced its basal and acquired thermotolerance, while overexpressing LlHsfA3B just enhanced its acquired thermotolerance48. Furthermore, AtHsfA3 was remarkably promoted in AtDREB2A CA and AtDREB2C overexpression seedlings, and the downregulation of Hsp18.1-CI and Hsp25.3-P in dreb2a and dreb2c depends on AtHsfA349,50. Li et al. confirmed that the direct interaction between AtHsfA2 and AtHsfA3 through genetic analysis, gene expression analysis and yeast two-hybrid assays51. HsfA3 regulates expression of many heat-inducible genes in the transcriptional cascade downstream of the DREB2A stress-regulatory system and functions in acquisition of thermotolerance under the control of the DREB2A cascade52. In addition, in the Phoenix dactylifera, transcript accumulation of PdHsfA3 imparting protection from heat-induced ROS generation and boosting the antioxidative response53. HsfA3 might play a similar role in the regulatory network in response to high-temperature stress in S. pohuashanensis. By functioning as molecular chaperones, Hsps prevent protein denaturation and aggregation. Hsp70 is the most prominent Hsp molecule that is visible when environmental temperature increment or other stresses that damage proteins54. In general, the expression of the Hsp70 positively correlates with the ability to tolerate high-temperature stress55. In the event of stress, Hsp70 provides protection to the cells by refolding and disaggregating substrate proteins56. A group of sHsps protect photosynthetic machinery from denaturation. sHsp21 plays an essential role in the development of thermomemory. It is reported that, in the tomato, these SlHsp21 mostly involve in the protection of PSII under high-temperature stress57. Transgenic Arabidopsis plants overexpressing Hsp21 show high tolerance to HS58. Other HSFs, like WRKYs, are another regulatory factor in response to high-temperature stress. AtWRKY25 can increase the contents of HsfA2, HsfB1, HsfB2A, and Hsp100F in AtWRKY25 overexpression lines of A. thaliana, which significantly improves the high-temperature resistance of the plants59,60. New evidence suggests that other TFs except for HSFs, for example ATHB and ERFs were also regulating plant's thermotolerance61. ATHB6 is a specific TF of higher plants and one of the components of the early signal transduction pathway, participating in the ABA signaling pathway as a main specific switch62. Moreover, ATHB6, ATHB7, and ATHB12 may interact with each other by forming heterodimers, which can activate potential complexes in plants63. ERFs belong to the AP2/ERF superfamily, which regulate the growth, development, and stress response of plants throughout the life cycle64. ERFs are early response factors that influence the expression of jasmonic acid (JA), salicylic acid (SA), ethylene (ETH), H2O2, and other signals by affecting the expression of downstream negative regulators, which affects the expression of defense genes and assists plants to resist environmental stress65,66. The increased transcription of MaERF1 in bananas causes the accumulation of MaERF1 under high temperature, indicating that MaERF1 may play an important role in the activation of the heat defense system67. Transcriptional regulations would play important roles in the regulatory network of S. pohuashanensis in response to high-temperature stress.
Hyperthermia breaks the homeostasis of ROS in cells, and changes of membrane proteins and membrane lipids, resulting in increased cell permeability, loss of cell membrane integrity and electrolyte outflow68. In this study, it was found that the content of MDA in S. pohuashanensis leaves increased significantly under high temperature stress. MDA is often used as an important indicator to reflect membrane lipid peroxidation, and its concentration represents the intensity of membrane lipid peroxidation and the damage degree of membrane system69. In addition, we found that GLR2.2 and GLR2.8 were up-regulated (Supplementary Table S10). Glutamate receptor protein (GLR) is a plant injury receptor, which can induce the increase of intracellular Ca2+ concentration in plants, and convert this signal into intracellular signal response to injury stimulation to induce plant defense response70. The increase of MDA content and the up regulation of GLRS in leaves of S. pohuashanensis under high temperature stress indicated that the cell membrane structure of S. pohuashanensis was damaged and could not maintain the normal cell structure. In addition, the contents of Pro, soluble sugar and other osmoregulation substances in S. pohuashanensis leaves also increased significantly after high temperature stress, indicating that the permeability of S. pohuashanensis cells increased and the normal metabolic activities of cells were affected. Transmembrane transport is one of the cellular activities affected by high temperature stress in S. pohuashanensis. Members of the plant NPF family have the function of transporting nitrate and small peptides, and several other NPF substrates, e.g. indole acetic acid (IAA), ABA, JAs, GAs, have been identified in recent years71,72. Meanwhile, the early secretion pathway of leaf cells was affected under high temperature stress. The early secretory pathway is an important stage for quality control and sorting of proteins73. Protein vesicles are formed at the endoplasmic reticulum export sites (ERES) and coat protein complex II (COPII) after preliminary processing of proteins in endoplasmic reticulum (ER), and separated from ER38. The above results showed that the cellular structures of the leaves of S. pohuashanensis were also destroyed due to the change of osmotic potential, and the transport capacity of transporters and plasma membrane (PM) decreased. It was unable to synthesize enough hormones, ROS scavenging enzymes and other substances to metabolize toxic substances significantly to enhance the further resistance of S. pohuashanensis under high temperature stress.
However, there are still some mechanisms of action for the maintenance of protein homeostasis under high temperature stress. In this study, specific expressions of molecular chaperones, resistant proteins and other types of genes were found (Supplementary Table S10). High temperatures lead to the accumulation of unfolded proteins in plant cells, which must be regenerated or degraded by plants in order to maintain normal life activities. Molecular chaperones are a class of proteins that correctly fold, assemble, transport other proteins and mediate the degradation of misfolded proteins, reducing the damage caused by stress to cells74. For instance, Heat shock protein 70 (Hsp70), a molecular chaperone, plays an extremely important role in biotic and abiotic stresses, and its activity is regulated by DnaJ, which is another type of chaperone. DnaJ can enhance the affinity of Hsp70 with its substrates, promote the release of Hsp7075,76. Up-regulated expression of chaperone genes indicated that S. pohuashanensis could regulates its homeostasis and reduces its damage by high-temperature to recombining various proteins denatured under high-temperature stress. Furthermore, in this study, autophagy related gene ATG8C-like and universal stress protein gene USP-like were down-regulated, and resistance genes RGA2-like, RGA3 and vicilin gene AMP2-2 were up-regulated. Autophagy is an intracellular quality control system that removes non-functional proteins and damaged cell components, and this homeostatic pathway is important for the energy balance of plant cells77. Recent studies have shown that autophagy is involved in the response of plants to high-temperature stress, and the impaired autophagy function of A. thaliana and Solanum lycopersicum leads to the accumulation of aggregated proteins, resulting in the decline of their heat tolerance78,79. High-temperature significantly induced up-regulated expression of autophagy-related genes ATG genes SlATG5 and SLATG7 in tomato, which resisted high temperature injury by protecting membrane structure and photosynthetic system80. Besides, some resistance proteins e.g. resistance gene analogs (RGAs), vicilin and universal stress proteins(USPs) seem to play a role in maintaining protein homeostasis of S. pohuashanensis under high-temperature stress42,81.
ROS is produced quickly after the beginning of high temperature stress, and can be used as an early messenger to activate stress response, and plants acquire heat tolerance by activating ROS scavenging system82. In this study, the activity of ROS scavenging enzymes of S. pohuashanensis showed that the activities of POD, T-SOD and APX in leaves of S. pohuashanensis were decreased after 8 h at 43℃. RNA-seq results showed that genes related to ROS scavenging enzymes in DEGs, e.g. PER47, PER47-like, PERP7-like and PNC1-like were down-regulated (Supplementary Table S10), which was consistent with the results of physiological indicators. Melakeselam and Zhou found that high temperature decreased POD and SOD activities in rape leaves, but had little effect on CAT activity83. APXs and CATs are considered to be two kinds of ROS scavenging enzymes necessary for ROS detoxification23. Although no genes related to CAT synthesis and metabolism were found in DEGs in this study, the results of physiological indicators also showed that CAT activity was not affected by high-temperature stress. It is possible that the response of CATs to environmental stress is comprehensive rather than only response to heat stress. GSTU belongs to glutathione S-transferases (GSTs). They are involved in phytochrome phyA mediated photomorphogenesis and integration of plant hormone signals, and regulate all aspects of plant development by affecting glutathione pool84. Stress treatments were applied to different spike weights of Oryza sativa at different temperatures. The activities of POD, SOD and CAT in leaves increased with increasing treatment temperature below 29 °C. In some varieties, POD activity started to decrease at 32 °C, and all enzyme activities showed a substantial decrease when the temperature reached 39 °C85. The indicators of antioxidant capacity of the Asparagus schoberioides wild type began to decline under high temperature stress at 32 °C, while the good variety ‘981′ maintained high antioxidant activity and the heat tolerant variety ‘07–2′ showed enhanced antioxidant activity capacity, with a significantly higher increase in the activities of some ROS scavenging enzymes (POD, GSH-Px) activity was significantly higher than wild type and '981′ under 28 °C treatment, indicating an important correlation between ROS scavenging enzymes activity and heat tolerance in A.schoberioides86. The biomass accumulation and cellulose content of the double-gene transformed Arabidopsis were higher under salt stress when compared to the wild-type and single-gene transformed plants, obtained by transfecting PaSOD and RaAPX simultaneously. The cellulose content was 60–100% higher in the double-transformed and PaSOD-transformed strains and 40% higher in the RaAPX-transformed strains compared to the wild-type strain. In addition, PaSOD and RaAPX double-transformed Arabidopsis accumulated more phenolics compared to the wild type87. All these results indicate that the scavenging of excess ROS is an important aspect of plant response to heat stress. However, in a previous study by Peng Song et al. it was found that SOD, and then POD activities of 1-year-old seedlings of S. pohuashanensis under high temperature stress at 40 °C increased and then decreased34. Furthermore, the same results were obtained in our subsequent replicate experiments. A possible important reason for the inability of rowan trees to adapt to high temperature stress is their reduced ability to scavenge reactive oxygen species. In addition to the ROS scavenging system, detoxification also played an important role in the occurrence of high-temperature stress. The expression of CYP71AV8-like was up-regulated in the DEGs (Supplementary Table S10). It is known that CYP71AV8 can convert n-pentene to n-pentanone, and participate in the biosynthesis of sesquiterpene lactone and artemisinic acid in chicory88. CYP71AV8 belongs to cytochrome P450 (CYP450), which exists in the membrane of PM, mitochondria, Golgi apparatus, peroxisome, nuclear membrane and other organelles. It has a detoxification effect. It can usually metabolize fat soluble toxic substances into water-soluble substances, so that toxic substances can be discharged from the body. As a terminal oxygenase, CYP450 is also involved in steroid hormone synthesis and other processes in vivo89. In this study, 14 genes of CYP450 in DEGs were identified except for CYP71AV8-like (Supplementary Table S10), which indicates that CYP450 may play a role in detoxification of plant cells in S. pohuashanensis under high-temperature stress. These results suggest that the resistance of S. pohuashanensis to high-temperature stress may be caused by the cooperation of multiple defense mechanisms.
When plants are subjected to stress conditions, maintaining adequate energy metabolism is essential for effective adaptation to stress90. Thylakoid membrane and matrix fluidity in chloroplasts increase, leading to inactivation of various enzymes involved in photosynthesis and disruption of the membrane system under high-temperature stress91. High temperature stress destroys the catalytic enzymes of dark reactions at first, and then PS II of light reactions92. However, as stress progresses, the degree of thylakoid destruction is aggravated, and at the same time, the PSII located on the thylakoid membrane is destroyed and the opening of the PSII reaction center is reduced34. This is consistent with the results that the fluorescence kinetic parameters F0/Fm of the leaves of S. pohuashanensis did not decrease significantly before 2 h of high-temperature stress, while parameters e.g. PSII and NPQ began to decrease after 2 h of high-temperature stress. In addition, the chloroplast NADP reductase gene LFNR-like, malate dehydrogenase gene NADP-ME, and transketolase gene TKL in DEGs were downregulated (Supplementary Table S11), indicating that the dark reaction process of photosynthesis was affected under high temperature stress. In the respiration, cytochrome C oxidase genes cob, cox1, cox2 and cox3 involved in the respiratory electron transport chain were up-regulated, and mitochondrial chaperone gene BCS1-like, respiratory-related gene FTSH4 and pyruvate metabolism gene MPC2-like were down-regulated (Supplementary Table S11), indicating that the respiration of the leaf cells of S. pohuashanensis was affected. It is worth mentioning that glyceraldehyde-3 phosphate dehydrogenase (GAPDH) gene GPP2 in DEGs was down-regulated (Supplementary Table S11), a substance considered to protect cells from environmental heat stress93. Moreover, KEGG enrichment of DEGs showed that the genes related to sulfur and nitrogen metabolism were down-regulated, tryptophan metabolism, arginine biosynthesis, phenylalanine metabolism, histidine metabolism, arginine and Pro metabolism and tyrosine metabolism were down-regulated in amino acid metabolism, indicating that the synthesis process of protein was significantly affected under high-temperature stress. The above results showed that high-temperature stress had an adverse effect on both material and energy metabolism of S. pohuashanensis.
On the whole, the calcium signal transduction pathway, phosphatidyl inositol signaling pathway, MAPK signaling pathway, RLKs, and plant hormone signaling pathways play important roles in the response mechanism of S. pohuashanensis to high-temperature stress. When S. pohuashanensis was subjected to high-temperature stress, the signals were transmitted to cells through transduction pathways, which regulated the expression of TFs and key heat resistance genes. At first, high-temperature stress affected the PM and caused changes in cell membrane fluidity, which activated Ca2+ channels and initiated the heat shock signal transduction pathway. At the same time, high temperatures also significantly affected the activity of RLKs on the membrane. Phosphoinositide, as a messenger, induced the release of calcium from the cell, thus rapidly increasing the calcium concentration in the cytoplasm. The signal triggered by high temperatures promoted the combination of Ca2+ and CaM, and Ca2+-CaM further activated calcium-dependent protein kinase (CDPK) to induce MAPKs to transport signals to the nucleus, and finally induced the expression of HSFs and other heat-resistant genes. cAMP, PLD, and other signaling pathways activated by G protein also played an important role in intercellular signal transmission. IAA and GA might be important plant hormones involved in the regulation of high-temperature morphological formation in S. pohuashanensis, and other endogenous hormones such as ABA and SA might also be involved in signal transduction under high-temperature stress, as endogenous signal molecules. However, it was unclear how the signals transport from the MAPK, cAMP, and PLD pathways to plant hormone signaling pathways, and further research would be needed. As the most important regulator, HSFs played an important role in the transcriptional regulatory network of S. pohuashanensis in response to high-temperature stress. HsfA3, NAC, ATHB, ERF, and other TFs are the key components that participated in the response of S. pohuashanensis to high-temperature stress and played a transcriptional regulatory role by regulating downstream genes. As part of the response to high temperature, HSF transcription factors rapidly induce the expression of Hsps (Hsp70, sHsp) and other cell components dealing with denatured proteins were also induced by high-temperature stress. Then the change of cell osmotic potential leads to the destruction of cell structure, and increase the major of stress-responsive osmolytes in S. pohuashanensis including Pro, soluble-sugar and protein, which play roles in maintaining cell morphology and cellular ionic homeostasis. Autophagy may be an intracellular quality control system in S. pohuashanensis that removes nonfunctional proteins and damaged cell components. Cell wall modification had positive significance for S. pohuashanensis to resist high-temperature. S. pohuashanensis activated ROS production upon sensing high-temperature stress. ROS functions might be a signaling molecule for activating local response as well as for transmitting to distal parts. Accumulation of ROS in S. pohuashanensis activated HSFs, which in turn activated ROS scavenging and detoxifying enzymes like APX and SOD. However, high-temperature destroyed the ROS homeostasis of S. pohuanshanensis and decreased the expression and activity of ROS scavenging enzymes. Detoxification alleviated the toxic state of cells to a certain extent besides. In response to high-temperature stress, S. pohuashanensis might enhance its stress resistance through the cooperation of multiple defense mechanisms (Fig. 11).

Schematic representation of the response mechanism of S. pohuashanensis to high temperature stress (Refer to Nishad et al. (2020) and Wang et al. (2020))20,22. High-temperature stress causes damage to membrane proteins, denaturation and inactivation of various enzymes, and accumulation of reactive oxygen species. High-temperature stress changes membrane fluidity, which may be sensed by proteins, such as Ca2+ channels and receptor-like kinases, localized at the plasma membrane. Calcium signaling plays critical roles in sensing sudden changes in temperature and activating cascades of signaling, leading to the production of Hsps that keep protein-unfolding under control. HSFs are the transcription factors that read the activation of thermosensors and induce the expression of HSPs. HsfAs are activated by high-temperature, and they target downstream transcription factors (such as HsfA3) to induce the expression of heat stress-responsive genes (Hsp70, sHsp, and other chaperones), which are important for ROS scavenging and protein homeostasis. In addition, some TF genes, including ERFs, bZIPs and ATHBs also play a role in the molecular mechanism of high-temperature stress in S. pohuashanensis. Accumulation of ROS in plants activates HSFs, which in turn activate ROS scavenging and detoxifying enzymes like APX and SOD. ROS level occurs due to the change by production of antioxidants, osmolytes, and Hsps. The major stress-responsive osmolytes in plants include proline, soluble-sugar and protein which play roles in maintaining cellular ionic homeostasis.
Materials and methods
Plant materials and treatment
One-year-old grafted seedlings from three S. pohuashanensis clones were selected from the forest germplasm resources nursery of the National Forest Genetic Resources Platform (NFGR), Beijing University of Agriculture. Three clones were deemed three biological replicates. Each biological replicate contained 10 plants randomly. Seedlings were transferred to a greenhouse under natural light (relative humidity 60%–70%) on March 1st, 2019. Eight weeks later, 2 seedlings from each clones (with 7–8 compound leaves) were selected randomly, then 6 plants were divided into a high-temperature treatment group (HT) and a control group (CK), and placed in an artificial climate chamber (BIC-400; Boxun, Shanghai, China). Seedlings were pretreated with a constant of 16 h/25 °C light, 8 h/18 °C dark, 70% relative humidity, and 180 μmol·m-2·s-1 light intensity before high-temperature treatment. After 3 d of pretreatment, the HT group was subjected to high-temperature stress at 43 °C for 8 h under light conditions (other conditions unchanged), while the conditions of the CK group remained unchanged. One seedling for each clone was chosen from each group. Chlorophyll fluorescence parameters of HT and CK groups were measured while high temperature treatment at 0, 2, 4, 6 and 8 h. At the end of treatment, all leaves of the HT and CK were collected, frozen in liquid nitrogen, and stored at -80 °C for RNA extraction and physiological measuring.
Determination of chlorophyll fluorescence parameters
The leaves of HT group and CK group were collected at 0, 2, 4, 6, 8 h after high temperature treatment (the leaves were shaded 20 min before collection). Chlorophyll fluorescence parameters such as the maximum photochemical efficiency (Fv/Fm), effective photochemical quantum yield (Fv'/Fm'), actual photochemical efficiency of PSII(ΦPSII), photochemical quenching coefficient (qL) and non-photochemical quenching coefficient (NPQ and qN) were measured by PAM-2500 chlorophyll fluorometers (Walz, Nuremberg, Germany).
Determination of osmoregulation substance content
The leaves of HT group and CK group were collected at 0 and 8 h after high temperature treatment Osmoregulation Substances consisted of MDA, Pro, soluble sugar and soluble total protein in leaves of HT group and CK group were determined by quantitative Kit [Malondialdehyde (MDA) assay kit (TBA method), A003-1–1, Proline assay kit, A107-1–1, Plant soluble sugar content test kit, A45-1–1, The total protein assay kit (BCA method), A045-3–1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China]. The absorbance values of each index were determined by 754 UV–Vis spectrophotometer (automatic) (Shanghai Jinghua, Shanghai, China). The determinations were carried out according to the manufacturer's instructions, and each biological replicate was repeated 5 times.
Determination of ROS scavenging enzyme activity
Leaves of HT group and CK group were collected at 0 and 8 h after high temperature treatment. Changes of activities of POD, T-SOD, APX, CAT were also detected according to the instruction of matched test kit provided by Nanjing Jiangcheng Bioengineering Institute [Peroxidase assay kit, A084-3–1, Total Superoxide Dismutase (T-SOD) assay kit (Hydroxylamine method), A001-1–1, Ascorbate peroxidase (APX) test kit, A123-1–1, Catalase (CAT) assay kit (Visible light), A007-1–1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China]. The absorbance values of each index were determined by 754 UV–Vis spectrophotometer (automatic). The determinations were carried out according to the manufacturer's instructions, and each biological replicate was repeated 5 times.
Total RNA isolation, mRNA library construction, sequencing, and transcriptome assembly
RNA samples were extracted from the leaves of S. pohuashanensis after 8 h of high temperature stress and control conditions. Total RNA was isolated from the samples using a TransZol Up Plus RNA Kit (Cat ER501-01; Transgen, Beijing, China). The integrity and concentration of total RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a Thermo Scientific NanoDrop2000 (Thermo Fisher Scientific, Wilmington, DE, USA). Library preparation and sequencing experiments were performed in accordance with the standard procedure provided by Illumina. Sequencing was performed using an Illumina HiSeq2000 system (Illumina, San Diego, CA, USA) by Shanghai Biotechnology Corporation (Shanghai, China).
Seqtk online software (https://github.com/lh3/seqtk) was used to obtain clean reads after raw reads obtained from sequencing were filtered adaptor sequences in reads, bases with quality Q lower than 30 at the 3 'end, reads with length less than 25, and ribosome RNA reads of the species.. Clean reads from the six libraries were assembled de novo using CLC Genomics Workbench (Version 6.0.4) to produce the primary unigenes 94,95. These were then assembled for a second time using CAP3 online stitching software (http://doua.prabi.fr/software/cap3) to acquire the final unigene sequence set and the quality of transcriptome assembly was assessed using BUSCO (Version 3.1.0) based on python96,97. These final unigenes were used for further exploration of the transcriptome. The accession number of this project is PRJNA699178.
Expression profile and enrichment analysis of DEGs
Final unigenes with differential expressions between the HT and CK groups of S. pohuashanensis were detected using eXpress software (Version 1.5.1) using three replicates per group98. Fragments Per Kilobase of Exon Per Million Fragments Mapped (FPKM) was used to calculate the expression abundance of each assembled transcript. Fold-change was calculated according to the FPKM value. The q-value and p-value after correction were obtained using R package edgeR (https://git.bioconductor.org/packages/edgeR99). If the Fold-change was much greater than 2 and the q-value was lower than 0.05, the unigene was considered to be differentially expressed.
For annotation, the unigene sequences were searched against the NCBI non-redundant protein database (Nr) using BlastX (http://www.ncbi.nlm.nih.gov/BLAST). Gene Ontology (GO) enrichment analysis of DEGs was performed using Blast2GO (http://www.Blast2GO.com). GO terms with e-values less than 1e-5 were considered significantly enriched in DEGs. We used the Kyoto Encyclopedia of Genes and Genomes (KEGG) KAAS online pathway comparative analysis tools (KAAS, http://www.genome.jp/KEGG/kaas/) to test the statistical enrichment of DEGs in KEGG pathways100.
Validation of DEGs by qRT-PCR
Quantitative RT-PCR was carried out using a Bio-Rad CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA, USA). The specific primers of 20 DEGs were designed using Primer Premier software (version 6.24) and then synthesized by Beijing Ruiboxingke Biotechnology Corporation (Supplementary Table S12). qRT-PCR was carried out using the 2 × T5 Fast qPCR Mix (SYBR Green I) (TSE202; Tsingke, Beijing, China) according to the manufacturer’s protocol. The actin gene of S. pohuashanensis was used as an internal marker. A comparative Ct method (2-△ΔCt) of relative quantification was used to evaluate the quantitative variation. All quantitative PCRs for each gene preformed three biological replicates with three technical replicates. The RNA samples for quantitative RT-PCR were the same as those used for Illumina sequencing.
Statistical analysis
Microsoft Excel 2016 (Microsoft Corp., Redmond, WA, USA) and Statistical Product and Service Solutions v.16.0 (SPSS, Chicago, IL, USA) were used to analyze experimental data. Data for P-values were analyzed by Student’s t-test at a significance level of 0.05. The method of Student–Newman–Keuls test (P < 0.05) was used for variance analysis.
Ethics approval and consent to participate
This study including sample collection was conducted according to China's Biodiversity Conservation Strategy and Action Plan (2011–2030) (Index number: 000014672/2010–00714) and Seed Law of the People's Republic of China (2015 Revised Version), which permits use of biological resources to Chinese for scientific research purpose.
References
Abbo, S., Lev-Yadun, S. & Gopher, A. Plant domestication and crop evolution in the Near East: on events and processes. Crit. Rev. Plant Sci. 31, 241–257 (2012).
Castro, A. V. P., Brito, P. & González-Rodríguez, A. Changes in thermic limits and acclimation assessment for an alpine plant by chlorophyll fluorescence analysis: Fv/Fm vs Rfd. Photosynthetica 56, 527–536 (2017).
Zheng, G., Tian, B. & Li, W. Membrane lipid remodelling of Meconopsis racemosa after Its introduction into lowlands from an alpine environment. PLoS ONE 9, e106614 (2014).
Mani, M. S. Alpine plants Book reviews: ecology and phytogeography of high altitude plants of the Northwest Himalaya Introduction to high altitude botany. Science 206, 883–896 (1979).
Berry, J. A. & Bjorkman, O. Photosynthetic response and adaptation to temperature in higher plants. Annu. Rev. Plant Physiol. 31, 491–543 (2003).
Vo, S. T. K. & Johnson, E. A. Alpine plant life: functional plant ecology of high mountain ecosystems by Christian Koerner. Mt. Res. Dev. 21, 202–202 (2001).
IPCC. https://www.ipcc.ch/report/ar5/wg2/ (2019).
Grover, A., Mittal, D., Negi, M. & Lavania, D. Generating high temperature tolerant transgenic plants: achievements and challenges. Plant Sci. 205, 38–47 (2013).
Ding, W., Chen, Z., Chen, J. & DIng J, Wei J, ,. Studies oil cultivating technology of Trollius chinensis in Beijing plain area. Chin. Traditional Herbal Drugs 34, s1–s4 (2003).
Li, X., Ji, L. & Hua, Z. Exogenous abscisic acid 013 heat resistance of Rhododendron lapponicum in Qinling Mountain. Guizhou Agric. Sci. 46, 33–36 (2018).
Ruelland, E. & Zachowski, A. How plants sense temperature. Environ. Exp. Bot. 69, 225–232. https://doi.org/10.1016/j.envexpbot.2010.05.011 (2010).
Tang, Y. et al. Heat stress induces an aggregation of the light-harvesting cmplex of photosystem II in spinach plants. Plant Physiol. 143, 629–638 (2007).
Feroza, K. C., Rosa, M. R., Eduardo, B. & Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 90, 856–867 (2017).
Qian, Y., Ren, Q., Zhang, J. & Chen, L. Transcriptomic analysis of the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Gene 692, 68–78. https://doi.org/10.1016/j.gene.2018.12.062 (2019).
Sedaghatmehr, M. et al. A regulatory role of autophagy for resetting the memory of heat stress in plants. Plant Cell Environ. 42, 1054–1064 (2018).
Yang, G. Y. et al. Two novel WRKY genes from Juglans regia JrWRKY6 and JrWRKY53 are involved in abscisic acid-dependent stress responses. Biol. Plant. 61, 611–621 (2017).
Raja, V., Majeed, U., Kang, H.-S., Andrabi, K. & John, R. Abiotic stress: Interplay between ROS, hormones and MAPKs. Environ. Exp. Bot. 137, 142–157. https://doi.org/10.1016/j.envexpbot.2017.02.010 (2017).
Ohama, N., Sato, H., Shinozaki, K. & Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 22, 53–65. https://doi.org/10.1016/j.tplants.2016.08.015 (2016).
Haq, S. et al. Heat shock proteins: dynamic biomolecules to counter plant biotic and abiotic stresses. Int. J. Mol. Sci. 20, 5321 (2019).
Nishad, A. & Nandi, A. K. Recent advances in plant thermomemory. Plant Cell Rep. https://doi.org/10.1007/s00299-020-02604-1 (2020).
Park, C.-J. & Seo, Y.-S. Heat shock proteins: a review of the Molecular chaperones for plant lmmunity. Plant Pathol J 31, 323–333 (2015).
Wang, G., Bai, W. & Wang, S. Advances in research of signal transduction and transcriptional regulatory mechanism of plants in response to heat stress. Molecular Plant Breeding 1–21 (2020).
Baxter, A., Mittler, R. & Suzuki, N. ROS as key players in plant stress signalling. J. Exp. Bot. 65, 1229–1240. https://doi.org/10.1093/jxb/ert375 (2014).
Slama, I., Abdelly, C., Bouchereau, A., Flowers, T. & Savoure, A. Diversity, distribution and roles of osmoprotective compounds accumulated in halophytes under abiotic stress. Ann Botany 115, 433–447 (2015).
Malinovsky, F. G., Fangel, J. U. & Willats, W. G. T. The role of the cell wall in plant immunity. Front. Plant Sci. 5, 178–189. https://doi.org/10.3389/fpls.2014.00178 (2014).
Yin, Y. et al. Triterpenoids from fruits of Sorbus pohuashanensis inhibit acetaminopheninduced acute liver injury in mice. Biomed. Pharmacother. 109, 493–502 (2019).
Liu, C. et al. De novo transcriptomic analysis and development of EST-SSRs for Sorbus pohuashanensis (Hance) Hedl. PLoS ONE 12, 179–219. https://doi.org/10.1371/journal.pone.0179219 (2017).
Kai, K. et al. Extraction and purification of anthocyanins from Sorbus Pohuashanensis fruits. Current Top. Nutraceut. Res. 18, 319–324 (2019).
Du, W. Secondary structure and phylogenetic relationship on sevsral species in Sorbus L., Nanjing Forestry University, (2016).
Tang, W. et al. Molecular evidence for maternal origin of sorbus with pink fruits. For. Res. 29, 834–838 (2016).
Jin, S., Yang, H. & Wang, J. Study on seed germination characteristics of Sorbus pohuashanensis. J. Jilin Forest. Sci. Technol. 48, 1–34 (2019).
Xu, M. et al. Study on nutritive substances and medicinal components of Sorbus pohuashanensis. For. Res. 33, 154–160 (2020).
Wu, Y. et al. Development and evaluation of EST-SSR markers in Sorbus pohuashanensis (Hance) Hedl and their application to other Sorbus species. Trees 34, 1–13 (2020).
Peng, S. et al. Physiological adaptation of Sorbus pohuashanensis seedlings to heat stress. For. Res. 24, 602–608 (2011).
Liu, C., Zhang, Z., Guan, X., Pei, X. & Zheng, J. Cloning and expression analysis of heat shock protein 70 gene in Sorbus pohuashanensis. Mol. Plant Breed. 17, 6276–6286 (2019).
Zhang, Z., Pei, X., Lu, Y., Chang, J. & Zheng, J. Cloning and expression analysis on small heat shock protein 23.8 gene(SpHSP23.8) in Sorbus pohuashanensis. J. Plant Resour. Environ. 29, 9–20 (2020).
Zhang, Z. et al. Molecular characterization and expression analysis of small heat shock protein 13 gene from Sorbus pohuashanensis (Hance) Hedl. in response to abiotic stress. Mol. Biol. Rep. 47, 1–11 (2020).
Tom´as, M., Mart´ınez-Alonso, E., Ballesta, J. & Mart´ınez-Men´arguez, J. A. Regulation of ER-Golgi intermediate compartment tubulation and mobility by COPI coats, motor proteins and microtubules. Traffic 11, 616–625. https://doi.org/10.1111/j.1600-0854.2010.01047.x (2010).
Reddy, A., Ali, G. S., Celesnik, H. & Day, I. S. Coping with stresses: roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 23, 2010–2032 (2011).
Majoul, T., Bancel, E., Triboi, E., Hamida, J. B. & Branlard, G. Proteomic analysis of the effect of heat stress on hexaploid wheat grain: characterization of heat-responsive proteins from total endosperm. Proteomics 3, 175–183 (2003).
Liu, H. et al. Contributions of PIP2-specific-phospholipase C and free salicylic acid to heat acclimation-induced thermotolerance in pea leaves. J. Plant Physiol. 163, 405–416 (2006).
Kim, D. J. et al. Crystal structure of the protein At3g01520, a eukaryotic universal stress protein-like protein from Arabidopsis thaliana in complex with AMP. Proteins: Struct., Funct., Bioinf. 83, 1368–1373. https://doi.org/10.1002/prot.24821 (2015).
Wang, T. et al. Phospholipase Dδ mediates the process of cold acclimation in Arabidopsis thaliana. Chin. Bulletin of Botany 45, 541–547 (2010).
Mishra, G., Zhang, W., Deng, F., Jian, Z. & Wang, X. A bifurcating pathway directs abscisic acid effects on stomatal closure and opening in Arabidopsis. Science 312, 264–266 (2006).
Von, K.-D.P., Dieter, S. K. & Lutz, N. The diversity of plant heat stress transcription factors. Trends Plant Sci. 12, 452–457 (2007).
Driedonks, N., Xu, J., Peters, J. L., Park, S. & Rieu, I. Multi-level iteractions between heat shock factors, heat shock proteins, and the redox system regulate acclimation to heat. Frontiers in Plant Science 6 (2015).
Kotak, S. et al. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 10, 310–316. https://doi.org/10.1016/j.pbi.2007.04.011 (2007).
Wu, Z. et al. Overexpression of two novel HsfA3s from Lily in Arabidopsis confer increased thermotolerance and salt sensitivity via alterations in proline catabolism. J. Exp. Bot. 69, 2005–2021 (2018).
Schramm, F. et al. A cascade of transcription factor DREB2A and heat stress transcription factor HSFA3 regulates the heat stress response of Arabidopsis. Plant J. 53, 264–274 (2008).
Chen, H. et al. Arabidopsis DREB2C functions as a transcription activator of HsfA3 during the heat stress response. Biochem. Biophys. Res. Commun. 401, 238–244 (2010).
Li, X. et al. Arabidopsis heat stress transcription factors A2 (HSFA2) and A3(HSFA3) function in the same heat regulation pathway. Acta Physiol. Plant. 39, 67–75 (2017).
Yoshida, T. et al. Functional analysis of an Arabidopsis heat-shock transcription factor HsfA3 in the transcriptional cascade downstream of the DREB2A stress-regulatory system. Biochem. Biophys. Res. Commun. 368, 515–521 (2008).
Khan, A. et al. Silicon and gibberellins: synergistic function in harnessing ABA signaling and heat stress tolerance in date palm (Phoenix dactylifera L.). Plants 9, 620–638 (2020).
Hahn, A., Bublak, D., Schleiff, E. & Scharf, K.-D. Crosstalk between Hsp90 and Hsp70 chaperones and heat stress transcription factors in tomato. Plant Cell 23, 741–755 (2011).
Finka, A., Mattoo, R. U. H. & Goloubinoff, P. Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones 16, 15–31. https://doi.org/10.1007/s12192-010-0216-8 (2011).
Bäurle, I. Plant heat adaptation: priming in response to heat stress. F1000 Res. 18, 1–5 (2016).
Neta-Sharir, I., Isaacson, T., Lurie, S. & Weiss, D. Dual role for tomato heat shock protein 21: protecting photosystem II from oxidative stress and promoting color changes during fruit maturation. Plant Cell 17, 1829–1838 (2005).
Charng, Y.-Y., Sinica, A., Liu, H.-C., Liu, N.-Y. & Chi, W.-T. A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol. 143, 251–262 (2007).
Li, S., Fu, Q., Huang, W. & Yu, D. Functional analysis of an Arabidopsis trancription factor WRKY25 in heat stress. Plant Cell Rep. 28, 683–693 (2009).
Li, S., Zhou, X., Chen, L., Huang, W. & Yu, D. Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Moleculer Cells 29, 475–483 (2010).
El-shershaby, A., Ullrich, S., Simm, S. & Scharf, K.-D. Functional diversification of tomato HsfA1 factors is based on DNA binding domain properties. Gene 714, 143985 (2019).
Cao, L., Ma, Y. & Yang, J. Plant transcript fctors of rsponse to aiotic sress Inner Mongolia Agricultural Science And Technology, 10–14 (2005).
Zhao, H., An, L. & Ma, Y. The progress of HD-Zip ATHB6. Chin. Agric. Sci. Bull. 22, 77–82 (2006).
Sun, B., Zhan, X., Cao, L. & Cheng, S. Research progress ofAP2/ERF transcription factor in rice (Oryza sativa). J. Agric. Biotechnol. 25, 1860–1869. https://doi.org/10.3969/j.issn.1674-7968.2017.11.013 (2017).
Chen, N. et al. Cloning of six ERF family transcription factor genes from peanut and analysis of their expression during abiotic stress. Plant Mol. Biol. Report. 30, 1415–1425. https://doi.org/10.1007/s11105-012-0456-0 (2012).
Liu, J. et al. Research progress of ERF transcription factors in plant biotic and abiotic stress responses. Acta Agric. Boreali-Sinica 28, 214–218 (2013).
Zhu, X., Wang, A., Zhu, S. & Zhang, L. Expression of ACO1, ERS1 and ERF1 genes in harvested bananas in relation to heat-induced defense against Colletotrichum musae. J. Plant Physiol. 168, 1634–1640. https://doi.org/10.1016/j.jplph.2011.03.009 (2011).
Zhang, Z., Min, H.-M., Xia, G.-J. & Jia, G.-M. Research advances on influence of high temperature stress on some physiological characteristics of plants. J. Anhui AgIi. Sci. 38, 8338–8339 (2010).
Luo, Q. et al. Physiological indices changes of Rhododendron simsii Planch. under high temperature stress and its heat resistance research progress. Modern Agric. Sci. Technol. 129, 125–126 (2018).
Toyota, M. et al. Glutamate triggers long-distance, calcium-based plant defense signaling. Science 361, 1112–1115 (2018).
Feng, H.-M., Lu, H., Wang, H.-Q. & Li, X.-Y. Function analyses of rice nitrate transporter gene OsNPF79 in nitrogen accumulation and transport. Chin. J. Rice. Sci. 31, 457–464. https://doi.org/10.16819/j.1001-7216.2017.7031 (2017).
Corratgé-Faillie, C. & Lacombe, B. Substrate (un)specificity of Arabidopsis NRT1/PTR FAMILY (NPF) proteins. J. Exp. Bot. 68, 3107–3113. https://doi.org/10.1093/jxb/erw499 (2017).
Wang, J., Luo, J. & Zhang, X. From endoplasmic reticulum to Golgi apparatus: a secretory pathway controlled by signal molecules. J. Zhejiang Univ. Med. Sci. 42, 472–477 (2013).
Hartl, F. U. & Hayer-Hartl, M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858 (2002).
So, H.-A., Chung, E. & Lee, J.-H. Molecular characterization of soybean GmDjp1 encoding a type III J-protein induced by abiotic stress. Genes Genom. 35, 247–256 (2013).
So, H.-A., Chung, E. & Lee, J.-H. Arabidopsis atDjC53 encoding a type III J-protein plays a negative role in heat shock tolerance. Genes Genom. 36, 733–744 (2014).
Liu, Y. & Bassham, D. C. Autophagy: pathways for self-eating in plant cells. Annu. Rev. Plant Biol. 63, 215–237 (2012).
Zhou, J. et al. NBR1-mediated selective autophagy targets insoluble ubiquitinated protein aggregates in plant stress responses. PLoS Genet. 9, e1003196 (2013).
Zhou, J., Wang, J., Yu, J.-Q. & Chen, Z.-X. Role and regulation of autophagy in heat stress responses of tomato plants. Front. Plant Sci. 30, 174 (2014).
Wang, J. Function,mechnism and mediation of tomato autophagy in resisitant to heat shock, Zhejiang University, (2013).
López, C. E. et al. Identifying resistance gene analogs associated with resistances to different pathogens in common bean. Phytopathology 93, 88–95 (2003).
Königshofer, H., Tromballa, H.-W. & Loeppert, H.-G. Early events in signalling high-temperature stress in tobacco BY2 cells involve alterations in membrane fluidity and enhanced hydrogen peroxide production. Plant Cell Environ. 31, 1771–1780 (2008).
Melakeselam, L. & Zhou, W. Effects of freezing and heat stresses on antioxidant enzyme activity, lipid peroxidation and membrane deterioration in rape. J. Zhejiang Agric. Univ. 25, 43–49 (1999).
Jiang, H.-W. et al. A glutathione S-transferase regulated by light and hormones participates in the modulation of Arabidopsis seedling development. Plant Physiol. 154, 1646–1658. https://doi.org/10.1104/pp.110.159152 (2010).
Zhu, X., Shao, J., Yang, W. & Ren, Z. Effects of temperature on protective enzymes activities and their isozymes expression in leaf of different panicle weight type of medium india hybrid rice. Acta Agric. Nucleatae Sinica 19, 260–264 (2005).
Lu, N. et al. Comparison of antioxidant activities of different strains of gracilaria lemaneiformis under high-temperature stress. J. Wuhan Univ. Nat. Sci. Ed. 56, 570–577 (2010).
Shafi, A. et al. Ectopic expression of SOD and APX genes in Arabidopsis alters metabolic pools and genes related to secondary cell wall cellulose biosynthesis and improve salt tolerance. Mol. Biol. Rep. 46, 1985–2002 (2019).
Cankar, K. et al. A chicory cytochrome P450 mono-oxygenase CYP71AV8 for the oxidation of (+)-valencene. FEBS Lett. 585, 178–182. https://doi.org/10.1016/j.febslet.2010.11.040 (2011).
Sauveplane, V. et al. Arabidopsis thaliana CYP77A4 is the first cytochrome P450 able to catalyze the epoxidation of free fatty acids in plants. FEBS J. 276, 719–735. https://doi.org/10.1111/j.1742-4658.2008.06819.x (2009).
Wang, Y., Luo, Z., Khan, Z., Mao, L. & Ying, T. Effect of nitric oxide on energy metabolism in postharvest banana fruit in response to chilling stress. Postharvest Biol. Technol. 108, 21–27 (2015).
Wang, J. Z., Cui, L. J., Wang, Y. & Li, L. J. Growth, lipid peroxidation and photosynthesis in two tall fescue cultivars differing in heat tolerance. Biol. Plant. 53, 237–242 (2009).
Wang, M. et al. Heat stress characteristic of photosystem IIin eggplant. Chin. J. Appl. Ecol. 18, 63–68 (2007).
Livramento, K. et al. Proteomic analysis of coffee grains exposed to different drying process. Food Chem. 221, 1874–1882 (2016).
Garg, R., Patel, R. K., Tyagi, A. K. & Jain, M. D. novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63 (2011).
Su, C. et al. De novo assembly of expressed transcripts and global analysis of the Phalaenopsis aphrodite transcriptome. Plant Cell Physiol. 52, 1501–1514 (2011).
Huang, X. & Madan, A. Cap3: a DNA sequence assembly program. Genome Res. 9, 868–877 (1999).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Roberts, A. Ambiguous fragment assignment for high-throughput sequencing experiments (University of California, 2013).
Robinson, M. D., Mccarthy, D. & Smyth, G. K. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Acknowledgements
We would like to thank the National Forest Genetic Resources Platform (NFGRP) for providing the S. pohuashanensis plant resources and the National Natural Science Foundation of China (Grant Number 31770369) for funding. We also thank Editage (www.editage.cn) for English language editing.
Author information
Authors and Affiliations
Contributions
X.P. performed the experiments and bioinformatics analysis, and drafted the manuscript; L.Z., D.Z., and Y.L. planted the sampling plants and analyzed the data; Y.Z. and J.Z. critically evaluated the protocol and data, and revised the final version of the manuscript. All authors read and approved the final manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pei, X., Zhang, Y., Zhu, L. et al. Physiological and transcriptomic analyses characterized high temperature stress response mechanisms in Sorbus pohuashanensis. Sci Rep 11, 10117 (2021). https://doi.org/10.1038/s41598-021-89418-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89418-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
