
Abstract
Amur ide (Leuciscus waleckii), a Cyprinid species, is broadly distributed in Northeast Asia. Different from its freshwater counterparts, the population in Lake Dali Nor has a strong alkalinity tolerance and can adapt to extremely alkali–saline water with bicarbonate over 50 mmol/L. To uncover the genetic basis of its alkaline adaptation, three populations, including one alkali form from Lake Dali Nor (DL), one freshwater form from its adjacent sister Lake Ganggeng Nor (GG), and one freshwater form from its historical origin, namely, the Songhua River (SH), were analyzed using genome resequencing technology. A total of 679.82 Gb clean data and 38,091,163 high-quality single-nucleotide polymorphism (SNP) loci were detected in the three populations. Nucleotide diversity and population structure analysis revealed that the DL and GG populations have lower nucleotide diversities and different genetic structures than those of the SH population. Selective sweeping showed 21 genes involved in osmoregulatory regulation (DLG1, VIPR1, AKT1, and GNAI1), inflammation and immune responses (DLG1, BRINP1, CTSL, TRAF6, AKT1, STAT3, GNAI1, SEC22b, and PSME4b), and cardiorespiratory development (TRAF6, PSME4b, STAT3, AKT1, and COL9A1) to be associated with alkaline adaption of the DL population. Interestingly, selective pressure (CodeML, MEME, and FEL) methods identified two functional codon sites of VIPR1 to be under positive selection in the DL population. The subsequent 3D protein modeling confirmed that these selected sites will incur changes in protein structure and function in the DL population. In brief, this study provides molecular evidence of population divergence and alkaline adaptation, which will be very useful for revealing the genetic basis of alkaline adaptation in Amur ide.
Similar content being viewed by others

Introduction
Amur ide (Leuciscus waleckii) belongs to Cyprinidae and is widely distributed throughout Northeast Asia. This species not only inhabits freshwater but also survives in alkali–saline water1. For example, Amur ide can inhabit Lake Dali Nor (116°25′–116°45′E, 43°13′–43°23′N), Inner Mongolia, China, which is a typical alkali–saline lake with HCO3−/CO32− concentrations greater than 50 mmol/L (pH 9.6) and salinities below 6‰2,3. In addition, the alkali Amur ide participates in spawning migration; it spawns in a small freshwater river (Shali River) in late April to early May every year and then returns to Lake Dali Nor for growth4,5. Geological and genetic studies have shown that the alkali form originated from the ancient freshwater forms of the Amur River during the early Holocene period6. Subsequently, due to the constant and harsh drought in the late Holocene period, Lake Dali Nor shrunk, and the water became seriously alkaline; Amur ide gradually adapted to the alkalinized environment and became the dominant fish species in Lake Dali Nor over the past several thousand years3,6.
The adaptation of Amur ide to the extreme alkali–saline environment occurs rapidly, which has represented a local adaptation pattern on a short evolutionary time scale of thousands of years. Previous studies have found that alkali form had stronger alkalinity tolerance and lower genetic diversity than its freshwater forms2,3. In recent years, using high-throughput sequencing technologies, transcriptomic expression profiles were compared in Amur ide ecotypes, revealing that many candidate genes associated with alkaline environments were differentially expressed1,2,4,8. Moreover, by comparing Amur ide in Lake Dali Nor and its adjacent sister freshwater form in Lake Ganggeng Nor, strong positive selection under alkaline environmental stress was observed for some genes1. Subsequently, by combining genome scans with landscape genomic methods, Xu, et al.9 found several genomic regions associated with alkaline adaptation under selective sweeps when comparing Amur ide in Lake Dali Nor and its ancestral freshwater form in the Amur River, which are candidate genes involved in processes such as ion homeostasis, reactive oxygen species elimination, and urea excretion. These findings suggest that Amur ide individuals dwelling in Lake Dali Nor have evolved unique genetic strategies that differ from their freshwater counterparts to cope with extremely alkaline environments.
Although several of the studies mentioned above reported observed phenotypic and genetic differentiation in Amur ide ecotypes, convincing evidence for the alkaline adaption of Amur ide is still lacking. Chang, et al.2 first analyzed six populations from the Amur River and Dali basin using microsatellite DNA markers and suggested that geographic isolation is likely the major force causing population divergence instead of a contrasting environment, demonstrating that it is necessary to combine historical origin and environmental factors to reveal the genetic basis of alkaline adaptation in Amur ide using abundant markers or genomic scans. Despite completely different environments, there are slight genetic differences due to gene flow between populations from Lake Dali Nor and its adjacent freshwater Lake Ganggeng Nor2,3. Therefore, to reduce or eliminate the discrepancy of spatial background and to focus on the differences of contrasting environments such as alkaline water and freshwater, we collected three populations, the alkali form from Lake Dali Nor (hereafter abbreviated as DL), the freshwater form from its adjacent sister Lake Ganggeng Nor (hereafter abbreviated as GG) and the freshwater form from its historical origin, the Songhua River (hereafter abbreviated as SH), which is one of the major branches of the Amur River (Fig. 1). Using high-throughput genome resequencing technology, the nucleotide diversity and population structure among the three populations were analyzed based on high-quality SNP calling data. Then, selective sweep analysis was performed to explore candidate genes for alkaline adaptation by comparing populations from contrasting environments. Finally, selective pressure (CodeML, MEME, and FEL) and protein structure analyses were applied to find evidence for adaptive selection in candidate genes from the DL population. This study aims to identify target genes or pathways related to alkaline adaptation and provides new insights into the genetic basis of alkaline adaptation of Amur ide.

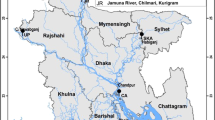
Geographical distribution of the sampled populations (solid red five-pointed stars represent the sampling locations). The map in the background has been generated by R package ‘ggmap’ (https://cran.r-project.org/web/packages/ggmap/)10.
Results
Resequencing genome profiles, SNP identification and nucleotide diversity
In this study, 679.82 Gb of clean data were collected, generating 48.56 Gb for each individual sample with 52.87-fold depth and 81.9% mapping rate on average to the reference genome of Amur ide (Supplementary Table S1). A total of 38,091,163 SNPs of high quality were obtained, including 12,610,411 in DL, 10,021,295 in GG, and 15,459,457 in SH. The SH population had the largest number of SNPs, and the difference was significant compared to the DL (two-tailed t-test, PSNP(SH/DL) = 2.53e−05**) and GG (PSNP(SH/GG) = 3.11e−05**) populations; while the two lake forms had no obvious differences in the number of SNPs (PSNP (DL/GG) = 0.74) (Fig. 2a). In addition, we detected 1290 population-specific SNPs in DL, 1334 in GG and 32,370 in SH, and the types of SNP substitutions in each population were counted (Fig. 2a). The largest numbers of substitution mutations were G > A in the DL and GG populations and T > C in the SH population (Supplementary Table S2). With annotation, we identified 7,718,559 SNPs, among which 4,893,256 are intergenic (63.40%), 2,548,902 are intronic (33.025%), and 276,401 are exonic (3.58%). Subsequently, SNPs in exonic regions were analyzed in detail, we identified 76,830 synonymous (26.40%), 126,782 nonsynonymous (43.58%), 59,763 3′UTR (20.54%), 13,026 5′UTR (4.48%), 5233 stop-lost (1.80%), 7122 stop-gained (2.45%) and 2183 unknown (0.75%) sites (Table 1, Fig. 2b).

SNP identification and nucleotide diversity in Amur ide ecotypes. (a) The number of SNPs (positive y-axis) and population-specific SNPs (negative y-axis) identified in each population. (b) Functional classification of the candidate SNPs. (aStop-gained: resulting in a premature stop codon in the coding sequence. bStop-lost: resulting in an elongated gene product because of stop codon loss.) (c) Violin plots of nucleotide diversity (π) for each population in 10-kb windows with10-kb steps. (d) LD decay estimated across the studied populations.
As Fig. 2c,d illustrated, SH had the highest nucleotide diversity and relatively fastest LD decay compared to DL and GG; GG had higher nucleotide diversity and relatively faster LD decay than those of DL; and DL had the lowest nucleotide diversity and the slowest LD level compared to the other two freshwater forms.
Population structure
After pruning 7,718,559 annotated SNPs for LD analysis (r2 > 0.4), approximate 19,760 SNPs in coding regions were extracted from the filtered 427,051 SNPs to examine the population structure and genetic relationships of the three populations. The ML tree showed that the two lake forms of DL and GG clustered together with small genetic distances (genetic distance < 0.001), whereas the SH population clustered into a single group, which showed a higher level of diversity and a larger genetic distance (genetic distance = 0.17 on average) than those of the DL and GG populations (Fig. 3a). This result was further supported by PCA, which showed that the SH population was more differentiated than the DL and GG populations (Fig. 3b). Furthermore, population structure analysis results based on STRUCTURE were also consistent with the ML tree and PCA results. When setting a cluster with K = 2, all populations converged to two clusters with the highest average likelihood value. The SH population exhibited fewer admixed and diversified genetic components than the other two populations (Fig. 3c).

Population structure in Amur ide ecotypes. (a) Maximum-likelihood phylogenetic tree. (b) Principal component analysis, PC 1 against PC 2. (c) Population structure. Colors in each fragment represent the proportion of K = 2 ancestral populations assigned to individual genomes.
Detection of selective candidate loci and genes associated with alkaline adaptation
Genome-wide annotated SNPs were utilized to calculate Fst and π ratio values of the two pairwise groups, which was performed with 10-kb window size and 10-kb step size. All windows containing less than 10 SNP sites were removed from the analysis. First, genomic loci with significantly high Fst values (PFst top 5% vs. 10-kb regions < 2.2e−16**) (0.1775 in GG/DL, 3258 windows and 0.375 in SH/DL, 3255 windows) and π ratios (Pπ ratios top 5% vs. 10-kb regions < 2.2e−16**) (3.709 in GG/DL, 3229 windows and 4.160 in SH/DL, 3259 windows) were identified as highly divergent loci (Fig. 4a and Fig. 4b).Then, 367 common loci containing 242 genes in GG/DL and 447 common loci containing 325 genes in SH/DL shared by both Fst and π ratio were detected. Finally, 51 common loci shared by two pairwise groups (GG/DL and SH/DL) were determined as candidate loci under positive selection of alkaline adaptation, with 21 genes being annotated (Table 2) (Supplementary Table S3). The strongest selective sweep signals were detected by comparing genomic regions under selective sweeps with the genome background, as shown by a box plot of the absolute difference in the Fst and π ratio (Fig. 4c). Among these selected genes, many are involved in immune responses and hypoxia-related pathways, including AKT1 (AKT serine/threonine kinase 1), STAT3 (signal transducer and activator of transcription 3), and DLG1 (discs large MAGUK scaffold protein 1) (Fig. 4d), of which AKT1 and STAT3 are enriched in the “Toll-like receptors signaling pathway” and “HIF-1 signaling pathway” and DLG1 in the “mitogen-activated protein kinase (MAPK) signaling pathway”.

Genomic regions under selective sweeps. (a,b) Distribution of the Fst and π ratio values, calculated in 10-kb windows with 10-kb sliding steps. Red data points were identified as selective sweeps that passed the thresholds of π ratio (the top 5% of the empirical distribution of π ratio, π ratio >= 3.709 in GG/DL and π ratio >= 4.160 in SH/DL) and Fst (the top 5% of the empirical distribution of Fst, Fst >= 0.1775 in GG/DL and Fst >= 0.375 in SH/DL). (a) The Fst and π ratio of GG/DL. (b) The Fst and π ratio of SH/DL. (c) Box plot of π ratio and Fst values for twenty-one genes versus the whole genome. (d) Representative common genes of two pairwise groups with strong selective sweep signals. Genome annotations are shown at the bottom, and black bars represent coding sequences.
Enrichment analysis of PSGs and intersection genes associated with alkaline adaptation
WEGO revealed that the top-ranked GO terms from each pairwise group shared by both the highest Fst and π ratio are related to local adaptations involving a range of biological processes, including biological regulation, cellular process, developmental process, metabolic process, pigmentation and response to stimulus (Fig. 5a). Further KEGG analysis showed that PSGs in the DL population cluster into several biological pathways, including autophagy, regulation of hypoxia-inducible factor (HIF) by oxygen, signaling by platelet-derived growth factor (PDGF), MAPK, relaxin signaling pathway, and metal ion transport (Fig. 5b) (Supplementary Table S4). Interestingly, some enrichment pathways that overlap with PSGs in the DL population were also found in gene enrichment analyses of stop-lost and stop-gained SNPs, such as the MAPK signaling pathway, chemokine and cytokine signaling pathway, PDGF signaling pathway, autophagy, HIF-1 signaling pathway, Toll-like receptor signaling pathway, and metal ion transport (Fig. 5c,d) (Supplementary Tables S5 and S6).

Enrichment analysis of candidate genes. (a) GO category analysis of genes using pairwise analysis. (b) KEGG category analysis of PSGs in the DL population. (c) KEGG category analysis of genes in stop-lost SNPs that are similar to PSGs. (d) KEGG category analysis of genes in stop-gained SNPs that are similar to PSGs.
Selective pressure analysis for candidate intersection genes associated with alkaline adaptation
The CodeML, MEME and FEL methods were used to detect the selective pressure of intersection genes among the PSGs and genes with stop-lost and stop-gained SNPs, and three genes, including VIPR1, DLG1, and GNAI1, were identified as being under selection (Fig. 6a). However, in addition to VIPR1, the site-based methods did not identify codons in DLG1 and GNAI1 under positive selective pressure. For VIPR1, positive selection of the 455th and 456th codons was found using the EasyCodeML and MEME methods, and positive selection of the 112th and 139th codons were detected in the MEME and FEL methods (Supplementary Tables S7 and S8). Among the four sites, only two (455th and 456th) are located in a well-defined protein domain (G protein-coupled receptors, GPCRs) (Fig. 6b). In addition, we examined the predicted 3D structures of VIPR1 in DL, GG and SH. The level of C-score confidence for three ecotypes were − 2.08, − 1.09, and − 1.99, respectively, indicating that the structures were constructed with high accuracy. The structural similarity and accuracy of the models were further checked using the TM-score, which showed all populations with TM-scores around 0.5 (0.47 ± 0.15, 0.58 ± 0.14, 0.48 ± 0.15, respectively), indicating that the modelled structure is of good quality. Interestingly, only the protein structure in DL has a wide and deep intracellular cavity in the core of VIPR1, which presumably forms a part of the peptide-binding site (Fig. 6c–e).

Target genes under selective pressure and differential VIPR1 protein structure in Amur ide ecotypes. (a) Venn diagram showing unique and overlapping genes among the PSGs and genes with stop-lost and stop-gained SNPs in DL, GG, and SH. (b) Structural analysis of the two positively selected sites of VIPR1. The protein coordinate is based on Ensembl ID ENSDARP00000116281. The lower panel shows the Pfam domains of the protein. The red stars indicate two sites (455th and 456th) that are essential for ligand binding. (c–e) Spatial distribution of positively selected sites in the 3D structure for Amur ide VIPR1. Three-dimensional views of VIPR1 proteins in DL (c), GG (d) and SH (e) highlight positively selected sites (455th and 456th) colored in the red rectangle frame.
Discussion
Effects of sample size
The number of samples and molecular markers are two important parameters to evaluate genetic diversity and differentiation of pupations. However, an increasing body of evidence showed that small sample sizes across thousands of SNPs can be highly informative for studying the genetic differentiation and relationships of populations11,12,13,14,15. Specifically, Nazareno et al.14 suggested that even two samples per population are adequate when ≥ 1500 SNPs are used. Furthermore, a study by Patterson et al.11 showed that approximately 10 individuals per population and ~1000 SNPs will be enough if the true Fst between two populations is 0.01. In the present study, despite only four to five samplers per population were used due to samples uneasy to obtained, a total of 19,760 SNPs were used for population structure analyses; combining to our previous results, the Fst values were 0.0949–0.1185 between Amur populations (including SH) and Dali populations (including DL and GG) using microsatellite markers2. Thus, the sample size used in this study is well within what is considered.
Nucleotide diversity and population structure
In this study, three parameters of SNP numbers, nucleotide diversity (π) and LD decay (r2) were used to evaluate nucleotide diversity of each population. According to our results, the freshwater form of SH had the largest number of SNPs, highest nucleotide diversity and relatively fastest LD decay, demonstrating that SH has increased levels of nucleotide diversity compared to DL and GG3,16. Relatively, the alkali form of DL had the lowest nucleotide diversity based on the values of the lowest nucleotide diversity and relatively slowest LD decay, which was mainly caused by increased inbreeding, limited gene flow and local adaptation inhabited in a lake without drainage as Chang et al.2 presumed before. Despite the freshwater form of GG likely experienced similar genetic events (e.g., genetic drift, bottleneck events, and inbreeding) as those that occurred in the DL population, it had moderate nucleotide diversity based on the values of higher nucleotide diversity and relatively faster LD decay compared to DL, the single direction of gene flow from DL to GG may explain for this based on the data reported by Chang et al.2
A clear population structure was found between the SH and the two lake populations of DL and GG (Fig. 3a–c). This is compatible with the geological evidence that the SH population was separated from populations from the Dali Basin and evolved into an independent population due to geographical isolation2. Furthermore, the two lake forms of the DL and GG were assigned to the same presumed population due to the similar genetic background2. However, the subsequent selective sweep analysis in our study identified some loci and genes associated with local adaptation in the alkaline environment of the DL population, implying that both geographical isolation and local adaptation caused the population divergence of Amur ide.
Screening for genes associated with alkaline adaptation
Despite a few studies have found genes associated with alkaline adaptation by comparing Amur ide ecotypes using selective sweeps, the differences of spatial background were no consideration. Xu et al.8 firstly reported the genes associated with alkaline adaptation by comparing DL and GG based on transcriptomic data without considering their genetic exchange; subsequently, Xu et al.9 identified genes associated with alkaline adaptation by comparing DL and its ancestral freshwater form from Amur river based on genomic data without considering spatial background differences (geographical isolation and water system like lake vs. river). Therefore, to eliminate spatial background differences interference (geographical isolation, water system, genetic exchange) and focus on contrasting environments alone (alkaline water and freshwater), we made two sets of selective sweep analyses (DL vs. GG and DL vs. SH) and took the intersection between the gene lists generated by each pairwise group identified regions, and identified genes related to alkaline adaptation with high credibility. Finally, a total of 51 genomic loci with 21 candidate genes were detected. We used GO and KEGG enrichment analyses to identify biological pathways overrepresented with these genes, and a range of biological processes related to immune responses, blood vessel development, and osmotic regulation were found, demonstrating that genomic scanning is a reasonable and effective way to determine genomic signatures of local adaptation. This also explains the reason for the reduced diversity of the DL population from the perspective of local adaptation7.
Stop-lost and stop-gained SNPs are nonsense variants that result in truncated or incomplete gene products17. To obtain comprehensive evidence of local adaptation, we assessed whether stop-lost and stop-gained SNPs are enriched in specific gene functions with respect to biological processes. Interestingly, a multitude of biological processes were overrepresented with genes containing stop-gained variants. Among them, biological processes related to local adaptation are of great interest. More importantly, most of them overlap with the biological pathways of PSGs, indicating that stop-lost and stop-gained SNPs may also have profound effects on the stress response to extremely alkaline environments.
Selective candidate genes with osmoregulatory regulation
Recent studies have shown that fish can sense changes in osmotic pressure in the external environment, and the pathways related to regulating osmotic pressure are transformed by sensory stimuli, thereby triggering many specific changes18,19. Phospholipase C (PLC) and MAPK signaling pathways are involved in osmotic pressure signaling in tilapia (Oreochromis mossambicus), killifish (Oryzias latipes) and turbot (Scophthalmus maxima)20,21.
In this study, we identified some genes (DLG1, VIPR1, AKT1, and GNAI1) and biological pathways (MAPK signaling pathway, relaxin signaling pathway, and metal ion transport) associated with mandatory physiological and structural alterations of osmoregulatory tissues20,22. The function of osmotic regulation affects a fish’s responses to extreme alkali–saline environments, and the genes and signaling pathways involved in this process can change the osmotic regulation strategies, which reverse the osmotic gradient between plasma/extracellular fluids and alkali–saline environments and complete the ion conversion between secretion and absorption.
There is no doubt that retention of water in the intestine occurs in response to alkaline and saline environments. VIPR1 belongs to G protein-coupled receptor class B1. In addition to vasoactive intestinal peptide (VIP), it binds to pituitary adenylate cyclase-activating polypeptide (PACAP), which is the most highly conserved member of the VIP-secretin-glucagon peptide superfamily. VIPR1 contributes to a variety of physiological functions, such as affecting the memory and learning system, stress response, neural development, immunomodulation, and exocrine secretion23,24,25,26,27. More importantly, it has been reported that VIPR1 acts on intestinal smooth muscle relaxation in mammals and promotes the discharge of water and electrolytes in the digestive system28,29. A few studies have reported that PACAP can relax smooth muscle in the stargazer by inhibiting contractions stimulated by acetylcholine or potassium chloride30, and recent evidence based on cryo-electron microscopy (cryp-EM) has further confirmed that PACAP27 (one form of PACAP) is embedded in the cavity of the VIPR1 protein structure 31. In the present study, VIPR1 is at the gene intersection among positive selection scanning, stop-gained, and stop-lost. Further 3D protein modeling showed that the opening of the hydrophobic receptor binding site in the cavity of VIPR1 is enlarged in the DL population, implying that VIPR1 in the DL population may be more effective at ligand binding than VIPR1 in the other two freshwater populations. Thus, we postulated that VIPR1 may play an important role in maintaining hydromineral balance during alkaline adaptation, which promotes smooth muscle relaxant actions by combining with PACAP in the digestive tract of the DL population.
Selective candidate genes associated with inflammation and immune responses
Recently, many studies have shown that some fish species living in harsh environments will stimulate many genes related to the immune system to fight against unfavorable habitats in the long term. Using comparative transcriptomics, Liang et al.32 found that a number of immune-related genes were triggered in the spleen of Amur carp (Cyprinus carpio haematopterus) at cooling temperatures. Tong et al.33 also reported that innate immune-related pathways were the most highly enriched in naked carp (Gymnocypris przewalskii), an alkali-saline-tolerant species inhabiting Lake Qinghai of China, to cope with the harsh living environment and pathogens (“white spot disease”).
In this study, we found that several genes (e.g., DLG1, BRINP1, CTSL, TRAF6, AKT1, STAT3, GNAI1, SEC22b, and PSME4b) and biological pathways (e.g., autophagy, Toll-like receptor signaling pathways, adaptive immune system, chemokine and cytokine signaling pathways, and signaling by interleukins) are associated with inflammation and immune responses, which might reflect the adaptation process of the DL population to the extreme alkali–saline environment. Apoptosis and tissue injury caused by the extreme environment cause damaged cells to release molecules that act as endogenous signals for the activation of inflammasome pathways and affect immune responses18. The immune system promotes the adaptation of the extreme alkali–saline environment by enhancing a variety of cytoprotective responses, thereby providing defense against inflammation and tissue damage caused by the environment. Moreover, the organism will activate the hypoxia signal pathway under hypoxic conditions and stimulate T cell differentiation and cytokine synthesis, thereby inhibiting the accumulation of inflammatory factors in the cell, repairing damaged blood vessels, and restoring the organism's balance34,35.
Selective candidate genes associated with cardiorespiratory development
Oxygen is the key factor for maintaining normal life activity and metabolism in fish. Some biotic and abiotic factors, such as temperature, ion concentration, pH, microorganisms, and algae, will change dissolved oxygen levels36,37,38, and some studies have indicated that the oxygen dissolution rate decreases with increasing water ion concentration39,40,41,42.
Considering the harsh environment with high alkalinity in Lake Dali Nor, inadequately dissolved oxygen is also a factor threatening the survival of the DL population35. In this study, we identified some genes (e.g., TRAF6, PSME4b, STAT3, AKT1, and COL9A1) and biological pathways (e.g., MAPK signaling pathway, HIF-1 signaling pathway, reactive oxygen species metabolic process, and signaling by PDGF) associated with cardiorespiratory development, indicating that the differentiation and development of blood vessels and cardiomyocytes play important roles in the DL population while living in a hypoxic environment43,44,45. Genes and signaling pathways involved in cardiorespiratory function can compensate for adverse effects caused by insufficient levels of dissolved oxygen in water, improve the efficiency of gas exchange in organisms, and maintain the stability of oxygen concentration46.
It is worth noting that several studies have focused on exploring HIF family genes related to hypoxia adaptation in fish. Eurasian perch (Perca fluviatilis) can upregulate expression of HIF-1a in the brain and liver under acute hypoxia conditions, while HIF-1a expression can change significantly in the muscle under chronic hypoxia conditions47. Indian catfish (Clarias batrachus) significantly upregulate three HIF-α (HIF-1α, HIF-2α, and HIF-3α) transcripts in the brain, liver, and head kidney under short-term hypoxia exposure; under long-term hypoxia exposure, HIF-1α in the spleen and HIF-2α in the muscle are significantly upregulated, and HIF-3α is downregulated in the head kidney48. Using genetic linkage analysis, Wang et al.49 identified HIF-3α as involved in alkaline adaptation of the DL population by suppressing gene expression in the gills. These findings implied that HIF family genes act as important modulators regulating angiogenesis. In addition, the VEGF signaling pathway regulates angiogenesis through increased expression of HIF-145, and interactions between the MAPK and VEGF signaling pathways can increase angiogenesis50.
Materials and methods
Ethics statement
In this study, all experiments involving the handling and treatment of fish were approved by the Animal Care and Use committee of Heilongjiang River Fisheries Research Institute of Chinese Academy of Fishery Sciences (HRFRI). The methods were carried out in accordance with approved guidelines. Before the blood samples were collected, all the fishes were euthanized in MS222 solution. In addition, we have followed the ARRIVE guidelines (http://www.nc3rs.org.uk/arrive-guidelines)51.
Sampling, DNA extraction, and resequencing
Three populations of Amur ide collected from Lake Dali Nor (DL), Lake Ganggeng Nor (GG) and the Songhua River (SH) were used in this study, including five individuals from DL, four individuals from GG, and five individuals from SH. Genomic DNA was extracted from each blood sample using a DNeasy Blood and Tissue Kit (Qiagen, Germany). The DNA concentration and integrity were evaluated using a NanoDrop 8000 (NanoDrop Technologies, USA) and 1% agarose gel electrophoresis. DNA libraries were prepared with greater than 1 μg of starting total DNA following Illumina protocols (Illumina Inc., USA), and then whole genome resequencing was completed on an Illumina HiSeq 4000 (Illumina Inc., USA) sequencing platform with a paired-end 150-bp strategy. The adaptors and low-quality bases (q < 20) were filtered out to obtain a set of clean paired reads by the FASTX-Toolkit (version 0.0.13) (http://hannonlab.cshl.edu/fastx_toolkit/).
SNP calling and annotation
The cleaned paired-end reads were mapped to the Amur ide reference genome (NCBI Accession Number GCA_900092035.1, Xu, et al. 9) using Bowtie2 (version 2.3.5)52. SNPs were called using the ‘mpileup’ command in SAMtools (version 1.9)53 and saved as bcf files (.bcf). All bcf files (.bcf) were then converted to vcf files (.vcf) with bcftools (version 1.9). In addition, we used bcftools to filter the vcf files. Unreliable SNPs that exhibited the following features were filtered out using bcftools: (1) coverage depth < 10 or > 1000; (2) root mean square (RMS) mapping quality < 20; and (3) read quality value < 20. Violin plots of the number of SNPs in each population were made with ggplot254 in the R package, and the significance levels were analyzed with a two-tailed Students t-test using R (version 3.6.3)55. Subsequently, we annotated and predicted the effects of the SNPs using snpEff (version 4.3)56. We constructed the necessary snpEff databases for Amur ide using GFF3 and FASTA files from reference assemblies of Xu et al.9. First, we obtained the publicly available Amur ide genome and annotation files, and renamed them as “genes.fa” and “genes.gff3”, respectively. Next, these two files were installed into the subfolder we created, the pre-existing “data” subfolder in the snpEff installation. We added a FASTA-formatted file containing the Amur ide genome reference sequences into the pre-existing “genome” subfolder in the above “data” folder, and genome annotation into the pre-existing "amur_ide" subfolder. We then added a new genome entry in the “snpEff.config” file in the snpEff directory: “amur_ide.genome: amur_ide”. Finally, we used the snpEff build command with parameters “-gff3 -v” to construct a custom snpEff database for Amur ide. Predicted effects of the detected SNPs were computed by the snpEff eff command with parameter option “-c”. The output file was then post-processed using a custom Python (version 3.6) script to isolate each component. Furthermore, population-specific SNPs were identified using the SnpSift component (version 4.1)57 in the snpEff java package.
Nucleotide diversity
We investigated the nucleotide diversity (π) for each population using VCFtools (version 0.1.13)58 in 10-kb non-overlapping windows (–window-pi 10000 –window-pi-step 10000). Prior to the LD decay analyses, the resulting filtered vcf files were converted to PLINK (.ped and .map) format file using VCFtools. The parameter r2 for LD was calculated using PLINK with the parameters (–ld-window -r2 0 –ld-window 99999 –ld-window-kb 50). The average r2 value was calculated for each length of distance and plotted against the physical distances of SNPs in units of kb. The corresponding figures were drawn by an R script55.
Population structure
All annotated SNPs were pruned using the indep-pairwise option (plink –file data –indep-pairwise 50 10 0.4) in PLINK (version 1.07)59 to avoid the strong influence of linked SNP clusters in relatedness analysis. We then extracted SNPs within the coding region in the filtered dataset for population structure and genetic relationships research. A maximum-likelihood (ML) tree was constructed based on the SNPs from the coding regions by using PhyML (HKY85 model) (version 3.1)60 and was plotted with iTOL (version 4.4.2). GCTA (version 1.91.1)61 was applied for principal component analysis (PCA). The Bayesian clustering program STRUCTURE (version 2.3.4)62 was used to analyze the distribution of SNPs among populations with 2000 iterations. StructurePlot (version 2.0)63 was performed to display the individual clusters of the estimated population structure.
Detection of selective signatures
To detect selection signatures associated with alkaline adaptation, we combined two approaches to select the positively selected genes (PSGs), including Fst and π ratio (πfreshwater/πalkaline water) of the freshwater forms of GG and SH to alkaline form of DL. First, we calculated the π ratio for DL, GG, and SH using VCFtools with a non-overlapping sliding window approach (10 kb windows with 10-kb stepwise distance, -window -pi 10000). The size and steps of the sliding window were based on empirical evaluation and referred to the practice of Xu et al.9. We separately estimated the π ratio (π_GG/π_DL and π_SH/π_DL). Secondly, we calculated the genome-wide distribution of Fst values using VCFtools with the same window size and stepwise distance (-fst -window -size 10000 -weir -fst -pop). Thirdly, loci from each pairwise group (GG/DL and SH/DL) with high Fst and π ratio values (corresponding to a top 5% level) were identified as selected loci. For each pairwise group, we compared Fst and π ratio values of top 5% SNP loci with those of 10-kb region by Students t-tests in R55 to determine significance. Finally, the selected loci shared by two pairwise groups were identified as highly divergent and PSGs were annotated. Furthermore, JBrowser (version 1.12.3)64 was used for the visualization of identified PSGs.
Enrichment analysis
Genes at selected loci from each pairwise group were classified into the GO database65 using WEGO (http://wego.genomics.org.cn/) and were annotated using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/) for the subsequent pathway analysis with the Kyoto Encyclopedia of Genes and Genomes (KEGG)66,67,68 and PANTHER69. Considering the drastic impact of selective SNPs on phenotypes17, genes in stop-lost and stop-gained SNPs from snpEff were also extracted and uploaded to KOBAS 3.0 for enrichment pathway analysis. P values were corrected by the False Discovery Rate (FDR) method of Benjamini–Hochberg and a significance threshold of FDR-corrected P < 0.05 was applied.
Selection pressure analysis for candidate intersection genes
We used the intersection among the PSGs and genes with stop-lost and stop-gained SNPs to conduct selection pressure analysis. First, the transcript sequences from the three Amur ide populations were retrieved. Then, the gene orthologs of six other representative outgroup fish species were obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank/); the accession numbers of the sequences were as follow: zebrafish (Danio rerio), XM_005162698; Nile tilapia (Oreochromis niloticus), XM_003439191; Mexican tetra (Astyanax mexicanus), XM_007249044; turquoise killifish (Nothobranchius furzeri), XM_015955164; Indian medaka (Oryzias melastigma), XM_024280491; and goldfish (Carassius auratus), XM_026200457. Next, these sequences were aligned using the ClustalW program (version 2.1)70, followed by manual adjustment. Finally, a phylogenetic tree was constructed using the ML method in the Mega X software71 with 1000 bootstrap replications. These output files were used in subsequent selective pressure analysis.
Three methods were used to perform selective pressure analysis. One was the CodeML method72 implemented in the EasyCodeML software (version 1.21)73. It was performed to calculate the nonsynonymous to synonymous substitution rate ratio (ω = dN/dS), where ω = 1, ω < 1 and ω > 1 correspond to neutral, purifying and positive selection, respectively74. A site-specific model was used to identify the variation of selective pressures on these genes, which allowed the ω ratio to vary among sites with a fixed ω ratio in all branches. Seven codon substitution models described as M0 (one-ratio), M1a (neutral), M2a (positive selection), M3 (discrete), M7 (β), M8 (β and ω > 1) and M8a (β and ω = 1) were investigated, and four different nested models (M0 vs. M3, M1a vs. M2a, M7 vs. M8 and M8a vs. M8) were calculated to detect positive selection on each site for candidate intersection genes75,76,77,78,79,80. The significance of differences between each two nested models was assessed using likelihood ratio tests (LRTs). Positively selected sites of genes with p < 0.05 in LRTs were further evaluated using Bayes Empirical Bayes (BEB)80 analysis with posterior probabilities ≥ 0.95. The other two methods were the mixed-effects model of evolution (MEME) and fixed effects likelihood (FEL) methods in the HyPhy package81 implemented on the Datamonkey server (http://www.datamonkey.org/). These methods were also used to infer the positively selected sites. Sites were considered candidates under positive selection when they met the following conditions: β + > α, significant likelihood ratio test (p < 0.05) in MEME, and p < 0.05 in the FEL likelihood ratio test. Sites identified by at least two methods were regarded as robust candidate positive selection sites.
Three-dimensional (3D) structure modeling of candidate intersection genes
To verify whether the positive selection sites obtained were located in important protein functional domains, the amino acid sequences of candidate genes from three Amur ide populations were aligned to corresponding zebrafish sequences to determine the equivalent positions of positively selected sites. Next, the Pfam webserver82 was used to determine whether the sites in zebrafish homologues have functional effects or are in proximity to functionally annotated sites. Then, the I-TASSER webserver (https://zhanglab.ccmb.med.umich.edu/I-TASSER/)83,84 was used to predict the 3D structure of the candidate genes and the structure was evaluated by two scoring function C-score and TM-score. For C-score, a good predicted model was obtained from a protein sequence when the estimated level of confidence was between [−5-2]. For TM-score, a model with TM-score > 0.5 indicates correct topology, and < 0.17 means the predicted structure with low accuracy85. Finally, the resulting model was visualized with PyMOL (http://pymol.sourceforge.net/).
Conclusion
Amur ide provides an excellent model for understanding the genetic basis of alkaline adaptation evolutionarily from the genomic level. Here, in order to eliminated spatial background differences interference (geographical isolation, water system, genetic exchange) and fucus on contrasting environments alone (alkaline water and freshwater), we re-sequenced genomes of three populations of Amur ide inhabiting different environments and identified 21 functional genes related to alkaline adaptation using selective sweeps based on a larger number of SNPs. Enrichment analysis showed that these genes mainly involved in osmoregulatory regulation, inflammation and immune responses, and cardiorespiratory development, which probably played important roles for Amur ide during alkaline adaptation. Further experiments are required to establish cause-and-effect relationships between phenotype and genotype. In summary, this study provides useful data for clarifying the genetic mechanisms of alkaline adaptation of Amur ide in the future.
References
Xu, J. et al. Gene expression changes leading extreme alkaline tolerance in Amur ide (Leuciscus waleckii) inhabiting soda lake. BMC Genom. 14, 682 (2013).
Chang, Y. M. et al. Genetic analysis of population differentiation and adaptation in Leuciscus waleckii. Genetica 141, 417–429 (2013).
Chi, B. J. et al. Genetic variability and genetic structure of Leuciscus waleckii Dybowski in Wusuli River and Dali Lake. J. Fish. Sci. China 17, 228–235 (2010) ((in Chinese with English abstract)).
Chen, B. H. et al. Transcriptional differences provide insight into environmental acclimatization in wild amur ide (Leuciscus waleckii) during spawning migration from alkalized lake to freshwater river. Genomics 111, 267–276 (2019).
Cui, J. et al. Transcriptional profiling reveals differential gene expression of amur ide (Leuciscus waleckii) during spawning migration. Int. J. Mol. Sci. 16, 13959–13972 (2015).
Geng, K. & Zhang, Z. C. Geomorphologic features and evolution of the Holocene lakes in Dali Nor Area, the Inner Mongolia. J. B. Normal. Univ. (Nat. Sci.) 4, 100 (1988) ((in Chinese with English abstract)).
Davis, C. D., Epps, C. W., Flitcroft, R. L. & Banks, M. A. Refining and defining riverscape genetics: How rivers influence population genetic structure. Wiley Interdiscip. Rev. Water 5, e1269 (2018).
Xu, J. et al. Transcriptome sequencing and analysis of wild amur ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS ONE 8, e59703 (2013).
Xu, J. et al. Genomic basis of adaptive evolution: The survival of amur ide (leuciscus waleckii) in an extremely alkaline environment. Mol. Biol. Evol. 34, 145–159 (2017).
Kahle, D. & Wickham, H. ggmap: spatial Visualization with ggplot2. The R J 5, 144–161 (2013).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Kotsakiozi, P. et al. Population genomics of the Asian tiger mosquito, Aedes albopictus: insights into the recent worldwide invasion. Ecol. Evol. 7, 10143–10157 (2017).
Willing, E.-M., Dreyer, C. & Van Oosterhout, C. Estimates of genetic differentiation measured by F ST do not necessarily require large sample sizes when using many SNP markers. PLoS ONE 7, e42649 (2012).
Nazareno, A. G., Bemmels, J. B., Dick, C. W. & Lohmann, L. G. Minimum sample sizes for population genomics: an empirical study from an Amazonian plant species. Mol. Ecol. Resour. 17, 1136–1147 (2017).
Flesch, E. P., Rotella, J. J., Thomson, J. M., Graves, T. A. & Garrott, R. A. Evaluating sample size to estimate genetic management metrics in the genomics era. Mol. Ecol. Resour. 18, 1077–1091 (2018).
Choi, J. W. et al. Genome-wide copy number variation in Hanwoo, Black Angus, and Holstein cattle. Mamm. Genome 24, 151–163 (2013).
Piot, A. et al. Genomic diversity evaluation of Populus trichocarpa germplasm for rare variant genetic association studies. Front. Genet. 10, 1384 (2020).
Kültz, D. Physiological mechanisms used by fish to cope with salinity stress. J. Exp. Biol. 218, 1907–1914 (2015).
Kültz, D. & Avila, K. Mitogen-activated protein kinases are in vivo transducers of osmosensory signals in fish gill cells. Comp. Biochem. Physiol. B: Biochem. Mol. Biol. 129, 821–829 (2001).
Loretz, C. A. et al. cDNA cloning and functional expression of a Ca2+-sensing receptor with truncated C-terminal tail from the Mozambique tilapia (Oreochromis mossambicus). J. Biol. Chem. 279, 53288–53297 (2004).
Marshall, W., Ossum, C. G. & Hoffmann, E. K. Hypotonic shock mediation by p38 MAPK, JNK, PKC, FAK, OSR1 and SPAK in osmosensing chloride secreting cells of killifish opercular epithelium. J. Exp. Biol. 208, 1063–1077 (2005).
Liu, C. et al. TAK1 promotes BMP4/Smad1 signaling via inhibition of erk MAPK: a new link in the FGF/BMP regulatory network. Differentiation 83, 210–219 (2012).
Ichikawa, S., Sreedharan, S. P., Owen, R. L. & Goetzl, E. J. Immunochemical localization of type I VIP receptor and NK-1-type substance P receptor in rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 268, L584–L588 (1995).
Ishihara, T., Shigemoto, R., Mori, K., Takahashi, K. & Nagata, S. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron 8, 811–819 (1992).
Kaltreider, H. B. et al. Upregulation of neuropeptides and neuropeptide receptors in a murine model of immune inflammation in lung parenchyma. Am. J. Respir. Cell Mol. Biol. 16, 133–144 (1997).
Reubi, J. C. In vitro evaluation of VIP/PACAP receptors in healthy and diseased human tissues: clinical implications. Ann. N. Y. Acad. Sci. 921, 1–25 (2000).
Reubi, J. C. et al. Vasoactive intestinal peptide/pituitary adenylate cyclase-activating peptide receptor subtypes in human tumors and their tissues of origin1. Cancer Res. 60, 3105–3112 (2000).
Lu, Y. & Owyang, C. Secretin-induced gastric relaxation is mediated by vasoactive intestinal polypeptide and prostaglandin pathways. Neurogastroenterol. Motil. 21, 754-e747 (2009).
Takei, Y. Exploring novel hormones essential for seawater adaptation in teleost fish. Gen. Comp. Endocrinol. 157, 3–13 (2008).
Matsuda, K. et al. Presence of pituitary adenylate cyclase-activating polypeptide (PACAP) and its relaxant activity in the rectum of a teleost, the stargazer, Uranoscopus japonicas. Peptides 21, 821–827 (2000).
Duan, J. et al. Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat. Commun. 11, 1–10 (2020).
Liang, L. Q., Chang, Y. M., He, X. L. & Tang, R. Transcriptome analysis to identify cold-responsive genes in amur carp (Cyprinus carpio haematopterus). PLoS ONE 10, e0130526 (2015).
Tong, C., Zhang, C. F., Zhang, R. Y. & Zhao, K. Transcriptome profiling analysis of naked carp (Gymnocypris przewalskii) provides insights into the immune-related genes in highland fish. Fish Shellfish Immunol. 46, 366–377 (2015).
Clambey, E. T. et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 109, E2784–E2793 (2012).
Scheinfeldt, L. B. & Tishkoff, S. A. Living the high life: high-altitude adaptation. Genome Biol. 11, 133 (2010).
Landsberg, J. H. The effects of harmful algal blooms on aquatic organisms. Rev. Fish. Sci. 10, 113–390 (2002).
Makrinos, D. L. & Bowden, T. J. Natural environmental impacts on teleost immune function. Fish Shellfish Immunol. 53, 50–57 (2016).
Stefan, H. G., Hondzo, M., Fang, X., Eaton, J. G. & McCormick, J. H. Simulated long term temperature and dissolved oxygen characteristics of lakes in the north-central United States and associated fish habitat limits. Limnol. Oceanogr. 41, 1124–1135 (1996).
Breitburg, D. Effects of hypoxia, and the balance between hypoxia and enrichment, on coastal fishes and fisheries. Estuaries 25, 767–781 (2002).
Long, Y. et al. Transcriptional events co-regulated by hypoxia and cold stresses in Zebrafish larvae. BMC Genom. 16, 385 (2015).
Thomas, P. & Rahman, M. S. Biomarkers of hypoxia exposure and reproductive function in Atlantic croaker: a review with some preliminary findings from the northern Gulf of Mexico hypoxic zone. J. Exp. Mar. Biol. Ecol. 381, S38–S50 (2009).
Zhu, C. D., Wang, Z. H. & Yan, B. Strategies for hypoxia adaptation in fish species: a review. J. Comp. Physiol. B. 183, 1005–1013 (2013).
Apte, R. S., Chen, D. S. & Ferrara, N. VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264 (2019).
Baptista, R. B., Souza-Castro, N. & Almeida-Val, V. M. F. Acute hypoxia up-regulates HIF-1α and VEGF mRNA levels in Amazon hypoxia-tolerant Oscar (Astronotus ocellatus). Fish Physiol. Biochem. 42, 1307–1318 (2016).
Semenza, G. L. HIF-1: using two hands to flip the angiogenic switch. Cancer Metastasis Rev. 19, 59–65 (2000).
Giordano, F. J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508 (2005).
Rimoldi, S. et al. HIF-1α mRNA levels in Eurasian perch (Perca fluviatilis) exposed to acute and chronic hypoxia. Mol. Biol. Rep. 39, 4009–4015 (2012).
Mohindra, V., Tripathi, R. K., Singh, R. K. & Lal, K. K. Molecular characterization and expression analysis of three hypoxia-inducible factor alpha subunits, HIF-1α,-2α and-3α in hypoxia-tolerant Indian catfish, Clarias batrachus [Linnaeus, 1758]. Mol. Biol. Rep. 40, 5805–5815 (2013).
Wang, N. et al. Screening microsatellite markers associated with alkaline tolerance in Leuciscus waleckii. J. Fish. Sci. China 022, 1105–1114 (2015) ((in Chinese with English abstract)).
Olsson, A. K., Dimberg, A., Kreuger, J. & Claesson-Welsh, L. VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 (2006).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412 (2010).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Wickham, H. ggplot2. WIREs Comput. Stat. 3, 180–185 (2011).
Team, R. C. R: A language and environment for statistical computing. (2013).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Ruden, D. M. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Baum, D. A., Small, R. L. & Wendel, J. F. Biogeography and floral evolution of Baobabs Adansonia, Bombacaceae as inferred from multiple data sets. Syst. Biol. 47, 181–207 (1998).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol. Ecol. Notes 7, 574–578 (2007).
Ramasamy, R. K., Ramasamy, S., Bindroo, B. B. & Naik, V. G. STRUCTURE PLOT: a program for drawing elegant STRUCTURE bar plots in user friendly interface. SpringerPlus 3, 1–3 (2014).
Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J. & Holmes, I. H. JBrowse: a next-generation genome browser. Genome Res. 19, 1630–1638 (2009).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. H. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucl. Acids Res. 38, W64–W70 (2010).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucl. Acids Res. 49, D545–D551 (2021).
Mi, H. Y., Muruganujan, A., Ebert, D., Huang, X. S. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucl. Acids Res. 47, D419–D426 (2019).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Yang, Z. H. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Gao, F. L. et al. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898 (2019).
Gao, F. et al. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898 (2019).
Anisimova, M., Bielawski, J. P. & Yang, Z. H. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 18, 1585–1592 (2001).
Suzuki, Y. & Nei, M. Reliabilities of parsimony-based and likelihood-based methods for detecting positive selection at single amino acid sites. Mol. Biol. Evol. 18, 2179–2185 (2001).
Swanson, W. J., Nielsen, R. & Yang, Q. F. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 20, 18–20 (2003).
Wong, W. S. W., Yang, Z. H., Goldman, N. & Nielsen, R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168, 1041–1051 (2004).
Yang, Z. H., Nielsen, R., Goldman, N. & Pedersen, A. M. K. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155, 431–449 (2000).
Yang, Z. H., Wong, W. S. & Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118 (2005).
Pond, S. L. K. & Muse, S. V. Statistical methods in molecular evolution 125–181 (Springer, Berlin, 2005).
Punta, M. et al. The Pfam protein families database. Nucl. Acids Res. 40, D290–D301 (2012).
Yang, J. Y. et al. The I-TASSER Suite: protein structure and function prediction. Nat. Meth. 12, 7–8 (2015).
Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 9, 40 (2008).
Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinf. 57, 702–710 (2004).
Funding
This research was funded by grants from National Key R & D Program of China (2019YFD0900405), National Natural Science Foundation of China (31602136); Natural Science Foundation of Heilongjiang Province of China (C2016070, ZD2018008), Central Public-interest Scientific Institution Basal Research Fund, CAFS (2019ZD0601, 2020TD22).
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.C.,Y.K. and S.W.; methodology, S.W., Y.K. and Y.C.; software, S.W. and Y.K.; formal analysis, S.W.; investigation, X.Z. and L.Z.; resources, L.L., B.S. and Y.C.; data curation, S.W.; writing—original draft preparation, S.W. and Y.C.; writing—review and editing, Y.C. and S.W.; visualization, S.W.; supervision, Y.C. and Y.K.; project administration, Y.C.; funding acquisition, L.L. and Y.C.; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, S., Kuang, Y., Liang, L. et al. Resequencing and SNP discovery of Amur ide (Leuciscus waleckii) provides insights into local adaptations to extreme environments. Sci Rep 11, 5064 (2021). https://doi.org/10.1038/s41598-021-84652-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84652-5
This article is cited by
-
Non-synonymous variation and protein structure of candidate genes associated with selection in farm and wild populations of turbot (Scophthalmus maximus)
Scientific Reports (2023)
-
Effects of chloride, sulfate, and bicarbonate stress on mortality rate, gill tissue morphology, and gene expression in mandarin fish (Siniperca chuatsi)
Environmental Science and Pollution Research (2023)
-
Variation in pH, HCO3−, carbonic anhydrases, and HCO3− transporters in Nile tilapia during carbonate alkalinity stress
Hydrobiologia (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
