
Abstract
Ulcerative colitis (UC) is one of the most common forms of inflammatory bowel disease (IBD) characterized by inflammation of the mucosal layer of the colon. Diagnosis of UC is based on clinical symptoms, and then confirmed based on endoscopic, histologic and laboratory findings. Feature selection and machine learning have been previously used for creating models to facilitate the diagnosis of certain diseases. In this work, we used a recently developed feature selection algorithm (DRPT) combined with a support vector machine (SVM) classifier to generate a model to discriminate between healthy subjects and subjects with UC based on the expression values of 32 genes in colon samples. We validated our model with an independent gene expression dataset of colonic samples from subjects in active and inactive periods of UC. Our model perfectly detected all active cases and had an average precision of 0.62 in the inactive cases. Compared with results reported in previous studies and a model generated by a recently published software for biomarker discovery using machine learning (BioDiscML), our final model for detecting UC shows better performance in terms of average precision.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gut with an increasing health burden1. Ulcerative colitis (UC) and Crohn’s disease are the two most common forms of chronic IBD with UC being more widespread than Crohn’s disease2. There is no cure for UC3 and people with the disease alternate between periods of remission (inactive) and active inflammation2. The underlying causes of UC are not completely understood yet, but it is thought to be a combination of genetic, environmental and psychological factors that disrupt the microbial ecosystem of the colon3,4. Genome-wide association studies (GWAS) have identified 240 risk loci for IBD5 and 47 risk loci specifically associated with UC6. However, the lower concordance rate in identical twins of 15% in UC compared with 30% in Crohn’s disease indicates that genetic contribution in UC is weaker than in Crohn’s disease7. Thus, using gene expression data for disease diagnostic might be more appropriate for UC than using GWAS data, as it has been done for Crohn’s disease8.
There are several features used for clinical diagnosis of UC including patient symptoms, and laboratory, endoscopic and histological findings7. Boland et at9 carried out a proof-of-concept study for using gene expression measurements from colon samples as a tool for clinical decision support in the treatment of UC. The purpose of Boland et al’s study was to discriminate between active and inactive UC cases; even though, they only considered gene expression of eight inflammatory genes instead of assessing the discriminatory power of many groups of genes, they concluded that mRNA analysis in UC is a feasible approach to measure quantitative response to therapy.
Machine learning-based models have a lot of potential to be incorporated into clinical practice10; specially in the area of medical image analysis11,12. Supervised machine learning has already proved to be useful in disease diagnosis and prognosis as well as personalized medicine13,14. In IBD, machine learning has been used to classify IBD paediatric patients using endoscopic and histological data15, to distinguish UC colonic samples from control and Crohn’s disease colonic samples16, and to discriminate between healthy subjects, UC patients, and Crohn’s disease patients using transcriptional profiles of peripheral blood17.
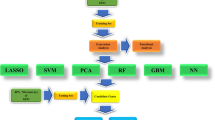
In this study, our goal was to investigate whether combining machine learning with a novel feature selection algorithm, an accurate model using the expression profiles of few (\(< 50\)) genes could be generated from transcriptome-wide gene expression data. To do this, we apply a machine learning classifier on gene expression data to generate a model to differentiate UC cases from controls. Unlike previous studies16,17, to reduce the effect of technical conditions, we combined a number of independent gene expression data sets instead of using a single data set to train our model. Additionally, by using feature selection, we were able to identify 32 genes out of thousands genes for which expression measurements were available. The expression values of these 32 genes is sufficient to generate a SVM model to effectively discriminate between UC cases and controls. On a gene expression dataset not used during training, our proposed model perfectly detected all active cases and had an average precision of 0.62 in the inactive cases.
Methods
Data gathering
We searched the NCBI Gene Expression Omnibus database (GEO) for expression profiling studies using colonic samples from UC subjects (in active and inactive state) and controls (healthy donors). We identified five datasets (accession numbers GSE115218,19, GSE1122320, GSE2261921, GSE7521422 and GSE945216). As healthy and Crohn’s disease subjects were used as controls in GSE945216, this data set was excluded from our study. We used three of the datasets for model selection using 5-fold cross-validation, and left one dataset for independent validation (Table 1). We partitioned the validation dataset into two datasets: Active UC vs controls, and inactive UC vs controls.
All data sets were obtained from studies where the diagnoses of patients were either based on endoscopical findings (GSE7521422 and GSE2261921), followed the criteria described by Lennard-Jones23 (GSE1122320), or based on clinical features as well as radiologic, endoscopic and laboratory findings (GSE115218). Disease state was either assessed during colonoscopy and classified into 1) no signs of inflammation (inactive), 2) low inflammation, and 3) moderate/high inflammation (active) (GSE22619); defined as active with a Mayo endoscopic sub-score \(\ge 2\) (GSE75214), or graded by a gastroenterologist or gastrointestinal pathologist (GSE11223, GSE1152). The control group had either normal mucosa at endoscopic level (GSE75214), no significant pathological findings during endoscopic and histological examinations (GSE22619), normal colonoscopies (GSE1152 and GSE11223) or tissues abnormalities other than IBD (GSE1152 and GSE11223).
For each dataset, GEO2R25 was used to retrieve the mapping between probe IDs and gene symbols. Probe IDs without a gene mapping were removed from further processing. Expression values for the mapped probe IDs were obtained using the Python package GEOparse26. The expression values obtained were as provided by the corresponding authors.
Data pre-processing
We performed the following steps for data pre-processing: (i) Calculating expression values per gene by taking the average of expression values of all probes mapped to the same gene. (i) Handling missing values with K-Nearest Neighbours (KNN) imputation method (KNNImputer) from the “missingpy” library in Python27. KNNImputer uses KNN to fill in missing values by utilizing the values from nearest neighbours. We set the number of neighbours to 2 (n-neighbours=2) and we used uniform weight.
To get our final training datasets we merged datasets GSE1152, GSE11223, and GSE22619 by taking the genes present in all of them. The merged dataset has 39 UC samples and 38 controls, and 16,313 genes. These same genes were selected from GSE75214 for validation. As the range of expression values across all datasets were different, we normalized the expression values of the final merged dataset and validation dataset by calculating Z-scores per sample.
Model generation
To create a model to discriminate between UC patients from healthy subjects, we selected the features (genes) using the dimension reduction through perturbation theory (DRPT) feature selection method28. Let \(D=[A\mid \mathbf{b} ]\) be a dataset where \(\mathbf{b}\) is the class label and A is an \(m\times n\) matrix with n columns (genes) and m rows (samples). There is only a limited number of genes that are associated with the disease, and as such, a majority of genes are considered irrelevant. DRPT considers the solution \(\mathbf{x}\) of the linear system \(A\mathbf{x} =\mathbf{b}\) with the smallest 2-norm. Hence, \(\mathbf{b}\) is a sum of \(x_i\mathbf {F}_i\) where \(\mathbf {F}_i\) is the i-th column of A. Then each component \(x_i\) of \(\mathbf{x}\) is viewed as an assigned weight to the feature \(\mathbf {F}_i\). So the bigger the \(|x_i|\) the more important \(\mathbf {F}_i\) is in connection with \(\mathbf{b}\). DRPT then filters out features whose weights are very small compared to the average of local maximums over \(|x_i|\)’s. After removing irrelevant features, DRPT uses perturbation theory to detect correlations between genes of the reduced dataset. Finally, the remaining genes are sorted based on their entropy.
Selected features were assessed using 5-fold cross-validation and support vector machines (SVMs) as the classifier. First, we performed DRPT 100 times on the training dataset to generate 100 subsets of features. Then, to find the best subsets, we performed 3 repetitions of stratified 5-fold cross-validation (CV) on the training dataset. We utilized average precision (AP) as calculated by the function average_precision_score from the Python library scikit-learn29 (version 0.22.1) as the evaluation metric to determine the best subset of genes among those 100 generated subsets. The four subsets with the highest mean AP over the cross-validation folds were chosen for creating the candidate models. For each of the four selected subset of features, we created a candidate SVM model using all samples in the training dataset. To generate the models, we used the SVM implementation available in the function SVC with parameter kernel=’linear’ from the Python library scikit-learn. To evaluate the prediction performance of each of the ten models, we validated it on the GSE75214-active and GSE75214-inactive datasets. In this step, we utilized the precision-recall curve (PRC) to assess the performance of the candidate models on unseen data. An additional candidate model was created using the most frequently selected genes.
BioDiscML
BioDiscML30 is a biomarker discovery software that uses machine learning methods to analyze biological datasets. To compare the prediction performance of our models with BioDiscML, we ran the software on our training dataset. 2/3 of the samples (N=52) were utilized for training and the remaining 1/3 (N=25) for testing. Since the software generates thousands of models, and we required only one model, we specified the number of best models as 1 in the config file (numberOfBestModels=1). One best model out of all models was created based on the 10-fold cross-validated Area Under Precision-Recall Curve (numberOfBestModelsSortingMetric= TRAIN-10CV-AUPRC) on the train set. We used Weka 3.831,32,33 to evaluate the performance of the model generated by BioDiscML, on the GSE75214-active and GSE75214-inactive datasets. Selected features by BioDiscML are C3orf36, ADAM30, SLS6A3, FEZF2, and GCNT3. In order to be able to use the model in Weka, we loaded the training dataset as it was created by BioDiscML, which was one of the outputs of the software. This dataset has six features, including selected genes and class labels, and 52 samples. We also modified our validation datasets by extracting BioDiscML selected features. After loading the training and test dataset in Weka explorer, we loaded the model, and we entered the classifier configuration as “weka.classifiers.misc.InputMappedClassifier -I -trim -W weka.classifiers.trees.RandomTree – -K 3 -M 1.0 -V 0.001 -S 1” which is the classifier’s set up in the generated model by BioDiscML.
Use of experimental animals, and human participants
This research did not involve human participants or experimental animals.
Results
Feature selection reduced significantly the number of genes required to construct a classification model
We performed DRPT 100 times on the training dataset to select 100 subsets of features. Then we performed 5-fold cross-validation to find the subsets with the highest mean average precision (AP) over the folds. The range of AP for the 100 subsets is between 0.82 and 0.97, with an average of \(0.91 \pm 0.03\). Table 2 shows the ten subsets with the highest cross-validated AP and the number of selected features (genes) on each subset. On average, DRPT selected \(37.55 \pm 8.84\) genes per subset.
Top five models are able to perfectly discriminate between active UC patients and controls

Identifying the most frequently selected genes. Top: Number of times each gene was selected. Genes were sorted based on the number of times they were selected by DRPT. Bottom: Normal QQ-plot. Horizontal line at 31 indicates the threshold selected to deem a gene as frequently chosen.
We selected the four top subsets with the highest mean AP, which are subsets 10, 51, 58, and 83 (Table 2), and created candidate models based on them. Each candidate model was created using all samples on the training dataset and the features of the corresponding subset. To identify the genes most relevant to discriminate between healthy and UC subjects, we looked at the number of times each gene was selected by DRPT. On 100 DRPT runs, 211 genes were selected at least once. The upper plot on Fig. 1 shows the number of times each gene was selected, and the lower plot shows the normal quantile-quantile (QQ) plot. Based on this plot, we saw that the observed distribution of the number of times a gene was selected deviates the most from a Gaussian distribution above 31 times. We considered the genes selected by DRPT more than 31 times as highly relevant and created a fifth model using 32 genes selected by DRPT at least 32 times over 100 runs.

Precision-recall curve of top selected subsets on GSE75214-active.

Precision-recall curve of top selected subsets on GSE75214-inactive.
In order to evaluate the prediction performance of the candidate models, each model was tested on the validation datasets, and PRC was plotted for model assessment (Figs. 2, and 3). As the AP approximates the AUPRC34, we used AP to summarize and compare the performance of these five models. All five candidate models achieved high predictive performance on the validation dataset GSE75214-active with an average AP of \(0.97 \pm 0.03\), while the average AP of these five models on the validation dataset GSE75214-inactive was \(0.60 \pm 0.06\). The models with the best performance were the model created with the 32 most frequently selected genes and subset 83 with an AP of 1 and 0.68 on GSE75214-active and GSE75214-inactive, respectively. However, based on a Friedman test35 (\(p-value=0.17\)), all five models have comparable performance on the validation datasets. We chose the model generated with the 32 most frequently selected genes as our final model.
Our top models outperformed the model generated by BioDiscML.
The average AUPRC achieved by the model created by BioDiscML on both GSE75214-active and GSE75214-inactive datasets was 0.798 and 0.544, respectively. Comparing the performance of our candidate models and the model created by BioDiscML on the two validation datasets, we observed that we achieved better AUPRC on both datasets (AUPRC = 1 on the active dataset, AUPRC = 0.68 on the inactive dataset). In terms of running time, subset selection by DRPT and final model creation and validation, took 3 minutes, while the running time of BioDiscML to create all the models and output the best final model was 1,890 minutes.
Links between the most frequently selected genes and UC.
We used Ensembl REST API (Version 11.0)36 to find the associated phenotypes with each gene belonging to the subset of the 32 most frequently selected genes (Table 3). Among these 32 genes, FAM118A is the only one with a known phenotypic association with IBD and its subtypes. The evidence supporting the association of some of the other 31 genes with UC based on phenotype is more indirect. For example, long term IBD patients are more susceptible to develop colorectal cancer37, and one of the 32 genes, TFRC, is associated with colorectal cancer. IBD patients are more prone to develop cardio vascular disease which is associated with blood pressure and cholesterol38, and four of the most frequently selected genes (LIPF, MMP2, DMTN and PPP1CB) are associated with blood pressure and cholesterol.
We looked at whether some of the 32 most frequently selected genes contained any of the 241 known IBD-associated SNPs5. To do this, we utilized Ensembl’s BioMart39 website (Ensembl Release version 98 - September 2019) to retrieve the genomic location of the 32 genes. We then used the intersectBed utility in BEDtools40 to find any overlap between the 241 IDB risk loci and the genomic location of the 32 genes. None of the IBD-associated SNPs was located on our 32 genes. Similarly, gene set enrichment analysis found no enriched GO term or pathway among these 32 genes. Additionally, these 32 genes are not listed as top differentially expressed genes in previous studies on UC41,42.
We searched the literature for links between the 32 genes and UC, and we found the following. MMP2 expression has been found significantly increased in colorectal neoplasia in a mouse model of UC43 and MMP2 levels are elevated in IBD44. TFRC has been found to have an anti-inflammatory effect on a murine colitis model45. KRT8 genetic variants have been observed in IBD patients and it was suggested that these variants are a risk factor for IBD46. DUOXA2 has been shown to be critical in the production of hydrogen peroxide within the colon and to be up-regulated in active UC47.
Discussion
In this study we showed the feasibility of using machine learning and feature selection to identify a reduced number of genes from microarray data to aid in the diagnosis of UC. One might argue that distinguishing UC patients from Crohn’s disease (CD) patients has more clinical relevance than distinguishing UC patients from controls. However, we were limited on the choice of groups to classify by data availability, as we could only find three gene expression data sets obtained from colonic samples of UC and CD patients in GEO (GSE1152, GSE75214 and GSE126124). As children samples were transcriptionally profiled for GSE12612448 instead of adults ones, we decided that the age difference could introduce extra biological variation in the expression data unrelated to UC. That left us with only two data sets which were not enough to train the model with multiple data sets and have at least one hold-out data set for validation.
Another limitation of this study is that we used gene expression profiles of colonic samples. Further research is required to assess the accuracy of our 32-gene model in gene expression profiles of blood samples. A recent study48 found a similar transcriptional profile between blood and colon tissue from patients with IBD. If indeed our 32-gene model is found accurate in blood samples, then a less invasive procedure such as a blood test could be used to diagnose UC instead of a colonoscopy or sigmoidoscopy.
In a previous study where machine learning was employed to perform a risk assessment for CD and UC using GWAS data49, a two-step feature selection strategy was used on a dataset containing 17,000 Crohn’s disease cases, 13,000 UC cases, and 22,000 controls with 178,822 SNPs. In that study, Wei et al reduced the number of features by filtering out SNPs with p-values greater than \(10^{-4}\) and then applied a penalized feature selection with \(L_1\) penalty to select a subset of SNPs. We decided against filtering out genes based on an arbitrary p-value of statistical significance of differential expression, as researchers are strongly advised against the use of p-values and statistical significance in relation to the null-hypothesis50,51.
Our 32-gene model achieved AP of 1 and 0.62 discriminating active UC patients from healthy donors, and inactive UC patients from healthy donors, respectively. We found direct or indirect links to UC for about a quarter of the 32 most frequently chosen genes. The remaining genes should be further investigated to find associations with UC. To put the performance of our 32-gene model into perspective, we looked at previous studies applying machine learning to create models for the diagnostic of UC. Maeda et al.52 extracted 312 features from endocystoscopy images to train a SVM to classify UC patients as active or healing. This approach achieve 90% precision at 74% recall; which is lower than the one achieved by our 32-gene model (Figs. 2, and 3). Yuan et al.17 applied incremental feature selection and a SMO classifier (a type of SVM) on gene expression data from blood samples to discriminate between healthy subjects, UC patients, and Crohn’s disease patients. The 10-fold cross-validation accuracy of their best model using the expression values of 1170 genes to classify UC patients was 92.31%, while our method obtained better accuracy than this with substantially less number of genes. In terms of potential for clinical translation of a machine learning-based model, a model requiring to quantify the gene expression levels of fewer genes is more suitable for the development of a new diagnostic test than one requiring the quantification of the expression levels of thousands of genes.
Using an efficient feature selection method such as DRPT and a SVM-classifier on gene expression data, we generated a model that could facilitate the diagnosis of UC from expression measurements of 32 genes from colonic samples. To avoid systematic experimental bias on the training data, we used three transcriptomic datasets from three separated studies. Our top model was validated with promising results on a data set not used for training; however, additional research is required to evaluate the 32 genes as potential biomarkers on a external set of subjects.
References
Kaplan, G. G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol.12, 720–727. https://doi.org/10.1038/nrgastro.2015.150 (2015).
Ordás, I., Eckmann, L., Talamini, M., Baumgart, D. C. & Sandborn, W. J. Ulcerative colitis. Lancet380, 1606–1619. https://doi.org/10.1016/S0140-6736(12)60150-0 (2012).
Eisenstein, M. Ulcerative colitis: towards remission. Nature563, S33. https://doi.org/10.1038/d41586-018-07276-2 (2018).
Khan, I. et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): cause or consequence? IBD treatment targeting the gut microbiome. Pathogens. https://doi.org/10.3390/pathogens8030126 (2019).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet.49, 256–261. https://doi.org/10.1038/ng.3760 (2017).
Anderson, C. A. et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet.43, 246–252. https://doi.org/10.1038/ng.764 (2011).
Conrad, K., Roggenbuck, D. & Laass, M. W. Diagnosis and classification of ulcerative colitis. Autoimmun. Rev.13, 463–436. https://doi.org/10.1016/j.autrev.2014.01.028 (2014).
Romagnoni, A. et al. Comparative performances of machine learning methods for classifying Crohn disease patients using genome-wide genotyping data. Sci. Rep.9, 10351. https://doi.org/10.1038/s41598-019-46649-z (2019).
Boland, B. S. et al. Validated gene expression biomarker analysis for biopsy-based clinical trials in ulcerative colitis. Aliment Pharmacol. Ther.40, 477–485. https://doi.org/10.1111/apt.12862 (2014).
Shah, P. et al. Artificial intelligence and machine learning in clinical development: a translational perspective. NPJ Digit. Med.2, 69. https://doi.org/10.1038/s41746-019-0148-3 (2019).
Esteva, A. et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature542, 115–118. https://doi.org/10.1038/nature21056 (2017).
McKinney, S. M. et al. International evaluation of an AI system for breast cancer screening. Nature577, 89–94. https://doi.org/10.1038/s41586-019-1799-6 (2020).
Molla, M., Waddell, M., Page, D. & Shavlik, J. Using machine learning to design and interpret gene-expression microarrays. AI Mag.25, 23 (2004).
Xu, J. et al. Translating cancer genomics into precision medicine with artificial intelligence: applications, challenges and future perspectives. Hum. Genet.138, 109–124 (2019).
Mossotto, E. et al. Classification of paediatric inflammatory bowel disease using machine learning. Sci. Rep.7, 2427. https://doi.org/10.1038/s41598-017-02606-2 (2017).
Olsen, J. et al. Diagnosis of ulcerative colitis before onset of inflammation by multivariate modeling of genome-wide gene expression data. Inflamm. Bowel Dis.15, 1032–1038. https://doi.org/10.1002/ibd.20879 (2009).
Yuan, F., Zhang, Y.-H., Kong, X.-Y. & Cai, Y.-D. Identification of candidate genes related to inflammatory bowel disease using minimum redundancy maximum relevance, incremental feature selection, and the shortest-path approach. Biomed. Res. Int.2017, 5741948. https://doi.org/10.1155/2017/5741948 (2017).
Moehle, C. et al. Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease. J. Mol. Med. (Berl)84, 1055–1066. https://doi.org/10.1007/s00109-006-0100-2 (2006).
Zahn, A. et al. Aquaporin-8 expression is reduced in ileum and induced in colon of patients with ulcerative colitis. World J. Gastroenterol.13, 1687 (2007).
Noble, C. L. et al. Regional variation in gene expression in the healthy colon is dysregulated in ulcerative colitis. Gut57, 1398–1405 (2008).
Lepage, P. et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology141, 227–236 (2011).
Vancamelbeke, M. et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm. Bowel Dis.23, 1718–1729 (2017).
Lennard-Jones, J. E. Classification of inflammatory bowel disease. Scand. J. Gastroenterol. Suppl.170, 2–6. https://doi.org/10.3109/00365528909091339 (1989) (discussion 16–9).
Häsler, R. et al. A functional methylome map of ulcerative colitis. Genome Res.22, 2130–2137 (2012).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res.41, D991–D995 (2012).
Gumienny, R. GEOparse. https://pypi.org/project/GEOparse/.
Troyanskaya, O. et al. Missing value estimation methods for DNA microarrays. Bioinformatics17, 520–525. https://doi.org/10.1093/bioinformatics/17.6.520 (2001).
Afshar, M. & Usefi, H. High-Dimensional Feature Selection for Genomics Datasets. Knowledge-Based Systems. https://arxiv.org/abs/2002.12104 (2020).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res.12, 2825–2830 (2011).
Leclercq, M. et al. Large-scale automatic feature selection for biomarker discovery in high-dimensional omics data. Front. Genet.10, 452 (2019).
Holmes, G., Donkin, A. & Witten, I. H. Weka: A machine learning workbench. In Proceedings of ANZIIS ’94 - Australian New Zealand Intelligent Information Systems Conference, 357–361 (1994).
Hall, M. et al. The weka data mining software: an update. ACM SIGKDD Explor. Newsl.11, 10–18 (2009).
Witten, I. H., Frank, E., Hall, M. A. & Pal, C. J. Data Mining: Practical Machine Learning Tools and Techniques (Morgan Kaufmann, Burlington, 2016).
Müller, A. C. et al.Introduction to Machine Learning with Python: A Guide for Data scientists (O'Reilly Media Inc, California, 2016).
Demšar, J. Statistical comparisons of classifiers over multiple data sets. J. Mach. Learn. Res.7, 1–30 (2006).
Yates, A. et al. The Ensembl REST API: Ensembl data for any language. Bioinformatics31, 143–145 (2014).
Kim, E. R. & Chang, D. K. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol.20, 9872 (2014).
Schulte, D. et al. Small dense LDL cholesterol in human subjects with different chronic inflammatory diseases. Nutr. Metab. Cardiovasc. Dis.28, 1100–1105 (2018).
Smedley, D. et al. Biomart-biological queries made easy. BMC Genom.10, 22 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics26, 841–842. https://doi.org/10.1093/bioinformatics/btq033 (2010).
Román, J. et al. Evaluation of responsive gene expression as a sensitive and specific biomarker in patients with ulcerative colitis. Inflamm. Bowel Dis.19, 221–229. https://doi.org/10.1002/ibd.23020 (2013).
Song, R. et al. Identification and analysis of key genes associated with ulcerative colitis based on DNA microarray data. Medicine (Baltimore)97, e10658. https://doi.org/10.1097/MD.0000000000010658 (2018).
Schwegmann, K. et al. Detection of early murine colorectal cancer by MMP-2/-9-guided fluorescence endoscopy. Inflamm. Bowel Dis.22, 82–91. https://doi.org/10.1097/MIB.0000000000000605 (2016).
Oliveira, L. G. D. et al. Positive correlation between disease activity index and matrix metalloproteinases activity in a rat model of colitis. Arq. Gastroenterol.51, 107–112. https://doi.org/10.1590/s0004-28032014000200007 (2014).
Shin, J.-S. et al. Anti-inflammatory effect of a standardized triterpenoid-rich fraction isolated from Rubus coreanus on dextran sodium sulfate-induced acute colitis in mice and LPS-induced macrophages. J. Ethnopharmacol.158(Pt A), 291–300. https://doi.org/10.1016/j.jep.2014.10.044 (2014).
Owens, D. W. & Lane, E. B. Keratin mutations and intestinal pathology. J. Pathol.204, 377–385. https://doi.org/10.1002/path.1646 (2004).
MacFie, T. S. et al. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm. Bowel Dis.20, 514–524. https://doi.org/10.1097/01.MIB.0000442012.45038.0e (2014).
Palmer, N. P. et al. Concordance between gene expression in peripheral whole blood and colonic tissue in children with inflammatory bowel disease. PLoS ONE14, e0222952. https://doi.org/10.1371/journal.pone.0222952 (2019).
Wei, Z. et al. Large sample size, wide variant spectrum, and advanced machine-learning technique boost risk prediction for inflammatory bowel disease. Am. J. Hum. Genet.92, 1008–1012 (2013).
Amrhein, V., Greenland, S. & McShane, B. Scientists rise up against statistical significance. Nature567, 305–307 (2019).
Wasserstein, R. L., Schirm, A. L. & Lazar, N. A. Moving to a world beyond “p< 0.05” (2019).
Maeda, Y. et al. Fully automated diagnostic system with artificial intelligence using endocytoscopy to identify the presence of histologic inflammation associated with ulcerative colitis (with video). Gastrointest. Endosc.89, 408–415. https://doi.org/10.1016/j.gie.2018.09.024 (2019).
Acknowledgements
This research was partially supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) to H.U. (Grant number RGPIN: 2019-05650) and to L.P.-C. (Grant number RGPIN: 2019-05247). H.M.K. was partially supported by funding from Memorial University’s School of Graduate Studies.
Author information
Authors and Affiliations
Contributions
Conceptualization H.U. and L.P.-C.; Methodology H.M.K., H.U. and L.P.-C.; Analysis H.M.K. and L.P.-C.; Writing H.M.K., H.U. and L.P.-C.; Experiments H.M.K.; Supervision H.U. and L.P.-C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khorasani, H.M., Usefi, H. & Peña-Castillo, L. Detecting ulcerative colitis from colon samples using efficient feature selection and machine learning. Sci Rep 10, 13744 (2020). https://doi.org/10.1038/s41598-020-70583-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70583-0
This article is cited by
-
Development of a 32-gene signature using machine learning for accurate prediction of inflammatory bowel disease
Cell Regeneration (2023)
-
Performance of Machine Learning Algorithms for Predicting Disease Activity in Inflammatory Bowel Disease
Inflammation (2023)
-
Identification of useful genes from multiple microarrays for ulcerative colitis diagnosis based on machine learning methods
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
