
Abstract
Zinc (Zn2+) can modulate platelet and coagulation activation pathways, including fibrin formation. Here, we studied the (patho)physiological consequences of abnormal platelet Zn2+ storage and release. To visualize Zn2+ storage in human and mouse platelets, the Zn2+ specific fluorescent dye FluoZin3 was used. In resting platelets, the dye transiently accumulated into distinct cytosolic puncta, which were lost upon platelet activation. Platelets isolated from Unc13d−/− mice, characterized by combined defects of α/δ granular release, showed a markedly impaired Zn2+ release upon activation. Platelets from Nbeal2−/− mice mimicking Gray platelet syndrome (GPS), characterized by primarily loss of the α-granule content, had strongly reduced Zn2+ levels, which was also confirmed in primary megakaryocytes. In human platelets isolated from patients with GPS, Hermansky-Pudlak Syndrome (HPS) and Storage Pool Disease (SPD) altered Zn2+ homeostasis was detected. In turbidity and flow based assays, platelet-dependent fibrin formation was impaired in both Nbeal2−/− and Unc13d−/− mice, and the impairment could be partially restored by extracellular Zn2+. Altogether, we conclude that the release of ionic Zn2+ store from secretory granules upon platelet activation contributes to the procoagulant role of Zn2+ in platelet-dependent fibrin formation.
Similar content being viewed by others


Introduction
Zinc (Zn2+) is an essential micronutrient, which modulates several enzymes, regulates the structure of zinc finger domains, induces diverse signaling pathways as a second messenger, and acts as an important cofactor in the metabolism1. Zn2+ circulates in the blood plasma at a concentration of 10–20 µM. However, only small amounts (0.1–2 µM) are present in the free ionic form, which can be taken up by platelets and other circulating blood cells2,3. Reduced Zn2+ uptake in the body results in altered platelet aggregation responses and impaired hemostasis, while intracellular chelation of Zn2+ in platelets inhibits tyrosine phosphorylation cascades as well as platelet reactivity and aggregation responses4,5,6. Interestingly, the Zn2+ concentration is considerably higher in platelets than in blood plasma7. Earlier findings showed that incubation with extracellular Zn2+ increases the Zn2+ concentration in the platelet cytoplasm and granules, pointing to the existence of Zn2+ uptake and storage mechanisms in these cells. Furthermore, the Zn2+ concentration in blood serum was found to be higher than in plasma, suggesting that activated platelets can release a significant amount of stored Zn2+ during clotting7. It has been suggested by some reports that protein-bound Zn2+ is accumulated in the platelet α-granules, which was explained by its high affinity for fibrinogen, albumin, histidine-rich glycoprotein, and factor XIII8. Extracellular Zn2+ directly binds fibrinogen and changes the fibrin fiber diameter, which is accompanied by increased clot stability9,10, raising the possibility that extracellular Zn2+ in the blood plasma, e.g. released from platelets, supports fibrin clot formation.
α-, δ- Storage pool disease (SPD) is characterized by deficiency of either α- or δ-granules or both types of granules in platelets. The Gray Platelet Syndrome in mouse11,12 and human13,14,15 is associated with an abolished gene function of Nbeal2 (NBEAL2), where primarily α-granule formation is severely impaired14,15. Mice lacking the Unc13d gene have an abolished δ-granule secretion and also partially defective exocytosis of α-granules and lysosomes16. These granular defects result in platelet dysfunction and severely prolonged tail-bleeding times in both, Unc13d−/− and Nbeal2−/− mice17.
In the present study, we show that the free ionic form of Zn2+ has a granular localization in both human and murine platelets, which is rapidly lost upon platelet activation. Characterization of Nbeal2−/− and Unc13d−/− strongly suggests that free zinc is mainly stored in α-granules. Further results highlight the importance of the platelet Zn2+ store and release in the modulation of coagulation and fibrin formation.
Results and Discussion
Given the unclearness of the location of intra-platelet ionic Zn2+ and the role of platelet Zn2+ release in clot formation, we re-evaluated these topics in human and mouse platelets. Loading of control human and WT mouse platelets with the Zn2+ specific fluorescence dye, FluoZin3, indicated that the cells of either species at resting state contained several stained foci, which became markedly reduced upon platelet spreading on a fibrinogen-coated surface. Some of the activated platelets became completely negative for FluoZin3 staining (Fig. 1A). This suggested that a substantial part of the intracellular free Zn2+ concentration ([Zn2+]i) in platelets is concentrated in granules, which are released upon activation. This was confirmed for FluoZin3-stained platelets in suspension measurement of the fluorescence intensity over time using flow cytometry (Fig. 1B). Addition of thrombin to the mouse platelet suspension caused a rapid and strong decrease in [Zn2+]i, (approximately 60–70%), (Fig. 1B). Taken together, this pointed to platelet secretory granules as major Zn2+ stores, although a remaining part of the Zn2+ can be available for Zn2+-binding proteins, such as metallothionein isoforms, as previously detected in megakaryocytes (MKs) and platelets18,19.

Distribution and levels of Zn2+ in human and mouse platelets. (A) Human (control) and mouse platelets loaded with FluoZin3 (green) were fixed (upper panels) or allowed to adhere on poly-lysine L (PLL) or spread on fibrinogen (FGN), (lower panels), stained with Atto 647N-phalloidin (red), and examined by confocal microscopy (representative images, Scale bar: 5 µm). (B) Washed WT, Unc13d−/− and Nbeal2−/− mouse platelets were loaded with FluoZin3, stimulated with thrombin (Thr-Activated, 0.1 U/mL), and fluorescence changes were observed by flow cytometry (MFI: mean fluorescence intensities). (C) Quantification of [Zn2+]i in mouse platelets, both resting and after thrombin stimulation (Thr-Activated); pre-incubation of WT, Unc13d−/− and Nbeal2−/− platelets with 100 μM ZnCl2 or 1 μM TPEN. Complete kinetic curve was recorded upon appending the first 50 sec (initial stage: Resting) to 400 sec (end point: Thr-Activated). Average of initial and end point measurements was shown. Each dot represents an individual mouse, Mean ± SEM. (D) In vitro differentiated bone-marrow megakaryocytes (MKs) from WT, Unc13d−/− and Nbeal2−/− mice were loaded with FluoZin3 (green), and then fixed (left panels) or allowed to adhere on poly-lysine L (PLL). Cells were examined by confocal microscopy (representative images); staining Atto 647N-phalloidin (red) and DAPI (blue). Each dote represents an independent experiment, n = 4 mice per group, Mean ± SEM. (E) Quantification of [Zn2+]i in FluoZin3-loaded MKs. Ratiometric analyses between FluoZin3 and phalloidin was shown. *P < 0.05; **P < 0.01; ***P < 0.001. 2-way ANOVA, Bonferroni’s multiple comparisons test, Student’s t-test.
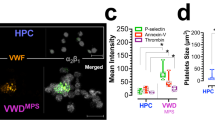
Release of the α- and δ-granule content from activated platelets affects several processes in the blood, including coagulation, wound repair and inflammation. Platelets from Unc13d−/− mice are characterized by an abolished δ-granule release and reduced α-granule secretion16. To study whether this defect in granule release is accompanied by a defect in Zn2+ efflux, we loaded platelets from both wild-type (WT) and Unc13d−/− mice with FluoZin3. In comparison to WT, platelets from Unc13d−/− mice showed a slightly reduced basal [Zn2+]i, but a severely impaired Zn2+ efflux after thrombin stimulation (Fig. 1A–C). Similarly, we used Nbeal2−/− mice, as a model of Gray Platelet Syndrome (GPS) lacking the α-granule content, which -in contrast to Unc13d−/− mice showed already under resting conditions severely reduced intracellular Zn2+ level, which upon activation were not much more reduced (Fig. 1A–C; Supplementary Fig. 1). To investigate further whether abnormal granular Zn2+ store of MKs could account for the dysregulated Zn2+ homeostasis in Nbeal2−/− platelets, bone marrow cells were differentiated to MKs in vitro. Subsequently, the FluoZin3 stained MKs were analyzed by confocal microscopy (Fig. 1D). Unc13d−/− MKs showed similar, homogenous FluoZin3 staining as WT MKs. However, abnormal accumulation of FluoZin3 staining close to the plasma membrane was observed in Nbeal2−/− MKs with strongly reduced fluorescence intensity within the cytoplasm (Fig. 1D,E). This suggested that an altered granular Zn2+ content in Nbeal2−/− MKs is a primary cause of the reduced [Zn2+]i in mutant platelets. To correlate our results to human platelet disorders, we isolated platelets from GPS13, HPS (Hermansky-Pudlak Syndrome), (Supplementary Table 1, Supplementary Fig. 2C) and SPD (Storage Pool Disease), (Supplementary Table 2) patients. Indeed, strongly reduced Zn2+ content was observed in resting and activated platelets from the GPS patient (Supplementary Fig. 2A) which resembles the observation made in Nbeal2−/− mice. Platelets isolated from the HPS patient had a reduced intracellular concentration of ionic Zn2+ under resting conditions and released lower amounts of Zn2+ compared to platelets from a healthy donor upon activation with thrombin. Similar observations were made in a number of patients with SPD (Supplementary Fig. 2B). Our preliminary results therefore suggest that Zn2+ homeostasis seems to be impaired in human platelets with granular abnormalities. Further investigations are necessary to confirm these results in a large cohort of patients.
To investigate further whether defective Zn2+ influx may contribute to the reduced [Zn2+]i in mutant platelets, we incubated WT, Unc13d−/− and Nbeal2−/− platelets with either ZnCl2 or the Zn2+ chelator TPEN as a negative control. We then measured [Zn2+]i concentrations again with FluoZin3. Even though the addition of ZnCl2 increased the basal [Zn2+]i in all mutant platelets, this level still did not reach that in WT platelets (Fig. 1C). This result is compatible with a regular Zn2+ uptake, but a defective storage in the mutant platelets. Markedly, TPEN treatment reduced the Zn2+ level in a similar manner in WT and Unc13d−/− platelets, but not in Nbeal2−/− platelets (Fig. 1C); suggesting that the free ionic Zn2+ store in Nbeal2−/− platelets is limited. Altogether, these alterations may point to an impaired Zn2+ transport into the (low numbers of) available secretory granules. In mammalians, several protein members of the ZIP protein family can mediate Zn2+ influx, thereby increasing [Zn2+]i concentrations. On the other hand, isoforms of the ZnT family regulate Zn2+ efflux from the cytosol to the extracellular space or into intracellular organelles lowering [Zn2+]i concentration1. Using in vitro grown primary MKs and quantitative RT-PCR, we confirmed mRNA expression of several ZIP/ZnT family members in this cell type (Supplementary Fig. 3). Whether the expression profiles of ZIP/ZnT transporters are altered in Nbeal2−/− and Unc13d−/− platelets, still needs to be investigated at mRNA and protein levels.
It is known that platelet-released Zn2+ can modulate local coagulant reactions, including contact activation and fibrin clotting9,10,20. Therefore, we considered that a defective granule biogenesis or granule secretion altering platelet Zn2+ release, may also affect the fibrin clotting process. First, a turbidity assay was performed to quantify the thrombin-induced fibrin formation according to the changes in the absorbance at 405 nm. WT, Nbeal2−/− and Unc13d−/− platelets were incubated in the presence or absence of extracellular Zn2+, prior to the activation with thrombin (Fig. 2A,B). We found that turbidity was lower in WT platelet-releasate, which was further decreased in the presence of extracellular Zn2+ (Fig. 2B). In Nbela2−/− mice turbidity was similar to resting level in platelet releasate after thrombin activation (Fig. 2A), but it was significantly reduced in the presence of Zn2+ (Fig. 2B). However, extracellular Zn2+ cannot fully restore fibrin formation to the WT level, due to the strongly reduced fibrinogen content and release from α-granules of Nbela2−/− platelets. No significant change was found in platelet releasate from Unc13d−/− mice neither in the presence nor in the absence of Zn2+, likely due to the abolished δ-granule and reduced α-granule secretion which strongly reduced fibrin clot formation in this experimental condition (Fig. 2B). To visualize fibrin formation, releasate of WT and mutant platelets were objected to scanning electron microscopy (SEM), and ultrastructure of fibrin clot was obviously different between WT, Unc13d−/− and Nbela2−/− platelets and the structure of fibrin clot was modified in the presence of Zn2+ (Supplementary Fig. 4A). However, further investigation is necessary to understand the exact role of platelet Zn2+ release in this process. Previous studies showed that 70% of α granules cannot be released in Unc13d−/− platelets due to the lack of ADP-mediated amplification of α-granule secretion16. To investigate ADP -dependent fibrin formation, turbidity assays were performed differently, in the absence (Supplementary Fig. 4B) or the presence (Supplementary Fig. 4C) of high dose of apyrase, stimulating platelet granule release with thrombin (1 U/mL). Similar results were obtained in these experimental settings as before, suggesting that Zn2+-induced fibrin formation does not require ADP at high dose of thrombin.

Effect of Zn2+ on fibrin formation under static and flow conditions in mouse models with defective α- and δ-granule biogenesis or secretion. (A,B) Turbidity assay on (A) resting and (B) thrombin-activated platelets. (B) WT, Unc13d−/− and Nbeal2−/− platelets were activated with 0.1 U/mL thrombin in the presence or absence of 100 μM ZnCl2, and turbidity was measured at 405 nm in an ELISA reader, n = 3 mice per group, Mean ± SEM. (C–H) Citrated whole-blood from WT, Unc13d−/− or Nbeal2−/− mice, labelled with DiOC6 (0.5 μg/mL) and AF647-fibrin(ogen), (8.5 μg/mL) in the presence or absence of 100 μM ZnCl2, was flowed under recalcification over a collagen surface for 8 min at a shear rate of 1,000 s−1. Platelet adhesion and fibrin formation were assessed by brightfield and multicolor fluorescence microscopy in time. Times to first fibrin formation recorded for individual flow runs. (C–H) Quantification of (C,E,G) fibrin and (D,F,H) adherent platelets. n = 6 mice per group, Mean ± SEM. SAC: surface adherent coverage. *P < 0.05; **P < 0.01; ***P < 0.001. 2-way ANOVA, Bonferroni’s multiple comparisons test, Student’s t-test.
Recording of the fibrin formation on platelet thrombi during whole-blood flow over collagen/tissue factor microspots provides an adequate way to evaluate hemostatic activity ex vivo21. Using microfluidics and a wall-shear rate of 1000 s−1, we assessed the kinetics of fibrin clot formation at such microspots from the accumulation rate of fluorescently-labeled platelets and fibrin (Supplementary Fig. 5). In blood samples from WT mice, Zn2+ addition did not change the kinetics of fibrin formation, thus indicating that Zn2+ was not a limiting factor in this process (Fig. 2C,D). Similar flow experiments were performed with blood from mice carrying the platelet secretion defects. Markedly, the platelet-dependent fibrin clot formation under flow was impaired in whole blood samples from Unc13d−/− (Fig. 2E,F) and Nbeal2−/− mice (Fig. 2G,H), but in both cases the kinetics of fibrin formation was accelerated by Zn2+ supplementation.
Altogether, our data significantly extend the early observation in 19851, that extracellular Zn2+ as well as platelet Zn2+ release have a procoagulant effect. In addition, our data indicate that several genetic dysorders impairing platelet granular content or release, especially in patients with GPS, could negatively influence platelet Zn2+-dependent fibrin formation and this defect could be partially rescued by Zn2+ supplementation. Our results also suggest that determination of the platelet Zn2+ content with FluoZin3 could be a novel prognostic biomarker for patients related storage pool disease and other bleeding disorders.
Material and Methods
Reagents
FluoZin3/AM (F24195, Invitrogen), N,N,N′,N′-tetrakis 2-pyridylmethyl ethylene diamine (TPEN) (ALX-450-011-M100, Enzo). Horm collagen (11207690, Nycomed), tissue factor (TF) was from Innovin (B4212-40, Dade-Behring, Marburg, Germany). Membrane dye DiOC6 was from AnaSpec. AF568-annexin A5 was from Life Technology (A13202, Eugene, USA) and AF647-labeled fibrinogen from Molecular Probes (F35200, Life Technology). Human fibrinogen (F4883) was from Sigma-Aldrich and thrombin from (10602400001) was from Roche Diagnostics. Phalloidin-AF647 (65906) was from Sigma-Aldrich. NucleoSpin®RNA II Kit was from Macherey-Nagel. High Capacity cDNA RT Kit was from Applied Biosystems and FastStart Universal SYBR Green Master Mix was from Roche. All reagents were bought from German suppliers unless otherwise stated.
Healthy subjects, patients and blood taking
All patients and blood donors were volunteers who gave free and informed written consent to participate in this study, conforming to the ethical standards adhering to the local Institutional Review Boards and the Declaration of Helsinki. Ethics committee of University of Würzburg Germany (reference: 52/15), and INSERM, France (reference: RBM-04-14) approved the study. Peripheral blood counts were within the reference ranges. SPD patients were diagnosed by standard routine diagnostics in the clinical centers showing the reported ADP/ATP content and release, as shown in Supplementary Table 2. The Gray Platelet Syndrome (GPS) patient was published earlier13 and Hermansky-Pudlak syndrome patient characterized by standard routine diagnostics and Transmission Electron Microscopy (TEM) in the respective clinical center, as shown in Supplementary Fig. 2C and Supplementary Table 1.
Animals
Unc13d−/− or Nbeal2−/− knockout mice have been previously described11,17. All experiments with animals were performed in accordance with the German legislation and guidelines of University of Würzburg and Regional Administration of Unterfranken (Lower District), Würzburg, Germany.
Confocal microscopy
For confocal microscopy, platelets (5 × 105/µL) were loaded with FluoZin-3/AM (5 µg/mL of dye in DMSO mixed 1:1 in 37 °C pre-heated pluronic acid), left shaking at 300 rpm; 30 min; 37 °C; dark. To allow the ester to cleave, loaded platelets are left idle for 20 min; 37 °C; dark. 125,000 of FluoZin3-AM loaded platelets/µL were fixed with 2% PFA-Phem, and allowed to adhere immediately on poly-L-lysine (for 20 min at room temperature), or allowed to spread on cover slips coated with fibrinogen (100 µg/mL) at 37 °C for 30 min after 0.01 U/mL thrombin stimulation (0.2 mM CaCl2 supplemented). The spread platelets were fixed with 2% PFA-Phem for 20 min. Adhered platelets were counter-stained with 100 µL of phalloidin-Atto 647 N (diluted 1:200 in PBS) for 1 h. Coverslips were washed with PBS, and mounted onto glass slides using Fluro shield mounting medium. Microscopic images were obtained using a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany), and analyzed with FiJi, Leica light and Imaris software.
Turbidity assay for fibrin formation
Washed platelets from WT, Unc13d−/− and Nbeal2−/− mice were activated with 0.1 U/mL or 1 U/mL thrombin in the presence or absence of 100 μM ZnCl2 or apyrase (2 U/mL) and were then incubated at 37 °C for 30 min. Thrombin-induced fibrin polymerisation was monitored for 2 hours by evaluating the turbidity at 405 nm using an ELISA reader.
Whole-blood fibrin formation on platelet thrombi in a flow system
Platelet-mediated fibrin formation in recalcified mouse whole blood using a microfluidics flow chamber with microspots of collagen and tissue factor, at a shear rate of 1000 s−1 was measured, as described before22.
Statistical analysis
Statistical significance was analyzed using SigmaPlot 11 software. Statistical difference of the means (Mean ± SEM) was determined using 2-way analysis of variance, followed by the stated test of variance for significance or an unpaired Student’s t test. P values < 0.05 were considered to be significant (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
References
Kambe, T., Tsuji, T., Hashimoto, A. & Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol Rev 95, 749–784 (2015).
Gorodetsky, R., Mou, X., Blankenfeld, A. & Marx, G. Platelet multielemental composition, lability, and subcellular localization. Am J Hematol 42, 278–283 (1993).
Whitehouse, R. C., Prasad, A. S., Rabbani, P. I. & Cossack, Z. T. Zinc in plasma, neutrophils, lymphocytes, and erythrocytes as determined by flameless atomic absorption spectrophotometry. Clin Chem 28, 475–480 (1982).
O’Dell, B. L. & Emery, M. Compromised zinc status in rats adversely affects calcium metabolism in platelets. J Nutr 121, 1763–1768 (1991).
Gordon, P. R., Woodruff, C. W., Anderson, H. L. & O’Dell, B. L. Effect of acute zinc deprivation on plasma zinc and platelet aggregation in adult males. Am J Clin Nutr 35, 113–119 (1982).
Watson, B. R. et al. Zinc is a transmembrane agonist that induces platelet activation in a tyrosine phosphorylation-dependent manner. Metallomics 8, 91–100 (2016).
Magneson, G. R., Puvathingal, J. M. & Ray, W. J. Jr. The concentrations of free Mg2+ and free Zn2+ in equine blood plasma. J Biol Chem 262, 11140–11148 (1987).
Marx, G., Korner, G., Mou, X. & Gorodetsky, R. Packaging zinc, fibrinogen, and factor XIII in platelet alpha-granules. J Cell Physiol 156, 437–442 (1993).
Henderson, S. J. et al. Zinc delays clot lysis by attenuating plasminogen activation and plasmin-mediated fibrin degradation. Thromb Haemost 113, 1278–1288 (2015).
Vu, T. T., Fredenburgh, J. C. & Weitz, J. I. Zinc: an important cofactor in haemostasis and thrombosis. Thromb Haemost 109, 421–430 (2013).
Deppermann, C. et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest 123, 3331–3342 (2013).
Guerrero, J. A. et al. Gray platelet syndrome: proinflammatory megakaryocytes and alpha-granule loss cause myelofibrosis and confer metastasis resistance in mice. Blood 124, 3624–3635 (2014).
Albers, C. A. et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet 43, 735–737 (2011).
Gunay-Aygun, M. et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet alpha-granules. Nat Genet 43, 732–734 (2011).
Nurden, A. T. & Nurden, P. Should any genetic defect affecting alpha-granules in platelets be classified as gray platelet syndrome? Am J Hematol 91, 714–718 (2016).
Harper, M. T., van den Bosch, M. T., Hers, I. & Poole, A. W. Platelet dense granule secretion defects may obscure alpha-granule secretion mechanisms: evidence from Munc13-4-deficient platelets. Blood 125, 3034–3036 (2015).
Stegner, D. et al. Munc13-4-mediated secretion is essential for infarct progression but not intracranial hemostasis in acute stroke. J Thromb Haemost 11, 1430–1433 (2013).
Rahman, M. T. & De Ley, M. Metallothionein in human thrombocyte precursors, CD61+ megakaryocytes. Cell Biol Toxicol 24, 19–25 (2008).
Sugiura, T. & Nakamura, H. Metallothionein in platelets. Int Arch Allergy Immunol 103, 341–348 (1994).
Marx, G. & Eldor, A. The procoagulant effect of zinc on fibrin clot formation. Am J Hematol 19, 151–159 (1985).
Swieringa, F., Kuijpers, M. J., Lamers, M. M., van der Meijden, P. E. & Heemskerk, J. W. Rate-limiting roles of the tenase complex of factors VIII and IX in platelet procoagulant activity and formation of platelet-fibrin thrombi under flow. Haematologica 100, 748–756 (2015).
Swieringa, F. et al. Platelet Control of Fibrin Distribution and Microelasticity in Thrombus Formation Under Flow. Arterioscler Thromb Vasc Biol 36, 692–699 (2016).
Acknowledgements
The authors thank Mike Friedrich from the Bioimaging center (Rudolf Virchow Center, Würzburg) for excellent technical support. This work was supported by grants from This study was supported by the Interreg Euregio Meuse Rhine programme PolyValve to M.N., S.B. and J.W.M.H.; and by the Deutsche Forschungsgemeinschaft project number 374031971-TRR 240/A09 to A.B. and H.M.H.
Author information
Authors and Affiliations
Contributions
S.K.G., J.P.G., M.N., E.M.B., J.V., L.W. and S.B. performed experiments and analyzed the data. C.D., J.E. and G.M. analyzed the data. B.N., S.E., U.S., C.S., P.N., A.G. and H.S (ORCID-ID: 0000-0003-1285-6407) provided essential tools and interpreted the data. A.B., H.M.H. and J.W.M.H. conceived and designed the research, interpreted the data and acquired funding. S.K.G., E.M.B., H.M.H., J.W.M.H. and A.B. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kiran Gotru, S., van Geffen, J.P., Nagy, M. et al. Defective Zn2+ homeostasis in mouse and human platelets with α- and δ-storage pool diseases. Sci Rep 9, 8333 (2019). https://doi.org/10.1038/s41598-019-44751-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44751-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
