
Abstract
Breviatea form a lineage of free living, unicellular protists, distantly related to animals and fungi1,2. This lineage emerged almost one billion years ago, when the oceanic oxygen content was low, and extant Breviatea have evolved or retained an anaerobic lifestyle3,4. Here we report the cultivation of Lenisia limosa, gen. et sp. nov., a newly discovered breviate colonized by relatives of animal-associated Arcobacter. Physiological experiments show that the association of L. limosa with Arcobacter is driven by the transfer of hydrogen and is mutualistic, providing benefits to both partners. With whole-genome sequencing and differential proteomics, we show that an experimentally observed fitness gain of L. limosa could be explained by the activity of a so far unknown type of NAD(P)H-accepting hydrogenase, which is expressed in the presence, but not in the absence, of Arcobacter. Differential proteomics further reveal that the presence of Lenisia stimulates expression of known ‘virulence’ factors by Arcobacter. These proteins typically enable colonization of animal cells during infection5, but may in the present case act for mutual benefit. Finally, re-investigation of two currently available transcriptomic data sets of other Breviatea4 reveals the presence and activity of related hydrogen-consuming Arcobacter, indicating that mutualistic interaction between these two groups of microbes might be pervasive. Our results support the notion that molecular mechanisms involved in virulence can also support mutualism6, as shown here for Arcobacter and Breviatea.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

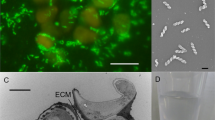
References
Brown, M. W. et al. Phylogenomics demonstrates that breviate flagellates are related to opisthokonts and apusomonads. Proc. R. Soc. B 280, 20131755 (2013)
Parfrey, L. W., Lahr, D. J. G., Knoll, A. H. & Katz, L. A. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl Acad. Sci. USA 108, 13624–13629 (2011)
Planavsky, N. J. et al. Low mid-Proterozoic atmospheric oxygen levels and the delayed rise of animals. Science 346, 635–638 (2014)
Stairs, C. W. et al. A SUF Fe-S cluster biogenesis system in the mitochondrion-related organelles of the anaerobic protist Pygsuia. Curr. Biol. 24, 1176–1186 (2014)
Ferreira, S., Queiroz, J. A., Oleastro, M. & Domingues, F. C. Insights in the pathogenesis and resistance of Arcobacter: a review. Crit. Rev. Microbiol. 42, 364–383 (2016)
Sayavedra, L. et al. Abundant toxin-related genes in the genomes of beneficial symbionts from deep-sea hydrothermal vent mussels. eLife 4, e07966 (2015)
McFall-Ngai, M. et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl Acad. Sci. USA 110, 3229–3236 (2013)
Suga, H. et al. The Capsaspora genome reveals a complex unicellular prehistory of animals. Nature Commun. 4, 2325 (2013)
Merchant, S. S. et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245–250 (2007)
Stairs, C. W., Leger, M. M. & Roger, A. J. Diversity and origins of anaerobic metabolism in mitochondria and related organelles. Phil. Trans. R. Soc. B 370, 20140326 (2015)
Schut, G. J. & Adams, M. W. W. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191, 4451–4457 (2009)
Hrdy, I. et al. Trichomonas hydrogenosomes contain the NADH dehydrogenase module of mitochondrial complex I. Nature 432, 618–622 (2004)
Stams, A. J. & Plugge, C. M. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nature Rev. Microbiol. 7, 568–577 (2009)
Moser, I., Schroeder, W. & Salnikow, J. Campylobacter jejuni major outer membrane protein and a 59-kDa protein are involved in binding to fibronectin and INT 407 cell membranes. FEMS Microbiol. Lett. 157, 233–238 (1997)
Monteville, M. R., Yoon, J. E. & Konkel, M. E. Maximal adherence and invasion of INT 407 cells by Campylobacter jejuni requires the CadF outer-membrane protein and microfilament reorganization. Microbiology 149, 153–165 (2003)
van Alphen, L. B. et al. Active migration into the subcellular space precedes Campylobacter jejuni invasion of epithelial cells. Cell. Microbiol. 10, 53–66 (2008)
Pernthaler, A., Pernthaler, J. & Amann, R. Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 68, 3094–3101 (2002)
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008)
Strous, M., Kraft, B., Bisdorf, R. & Tegetmeyer, H. E. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3, 410 (2012)
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012)
Nadalin, F., Vezzi, F. & Policriti, A. GapFiller: a de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13 (Suppl. 14), S8 (2012)
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008)
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O. & Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990 (2008)
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004)
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14 (2004)
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012)
Emanuelsson, O., Nielsen, H., Brunak, S. & von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 (2000)
Claros, M. G. MitoProt, a Macintosh application for studying mitochondrial proteins. Comput. Appl. Biosci. 11, 441–447 (1995)
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011)
Finn, R. D. et al. The Pfam protein families database. Nucleic Acids Res. 38, D211–D222 (2010)
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015)
Glass, E. M., Wilkening, J., Wilke, A. & Antonopoulos, D. & Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 5, http://dx.doi.org/10.1101/pdb.prot5368 (2010)
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. PeerJ 3, 1–4 (2014)
Smith, C. J., Nedwell, D. B., Dong, L. F. & Osborn, A. M. Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Appl. Environ. Microbiol. 73, 3612–3622 (2007)
Parfrey, L. W. et al. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst. Biol. 59, 518–533 (2010)
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012)
Winiewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nature Methods 6, 359–362 (2009)
Oberg, A. L. & Vitek, O. Statistical design of quantitative mass spectrometry-based proteomic experiments. J. Proteome Res. 8, 2144–2156 (2009)
Olsen, J. V. et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 (2005)
Spivak, M., Weston, J., Bottou, L., Käll, L. & Noble, W. S. Improvements to the percolator algorithm for peptide identification from shotgun proteomics data sets. J. Proteome Res. 8, 3737–3745 (2009)
Florens, L. et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303–311 (2006)
Mueller, R. S. et al. Ecological distribution and population physiology defined by proteomics in a natural microbial community. Mol. Syst. Biol. 6, 374 (2010)
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001)
Zhu, Y., Stephens, R. M., Meltzer, P. S. & Davis, S. R. SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics 14, 19 (2013)
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010)
Simpson, J. T. & Durbin, R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556 (2012)
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015)
Hung, Y. P., Albeck, J. G., Tantama, M. & Yellen, G. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 14, 545–554 (2011)
Acknowledgements
We thank T. Hargesheimer, D. Liu, G. Klockgether, P. Hach, R. Appel and I. Kattelmann for technical assistance, A. Simpson, G. Strous and Fulvio Reggiori for comments on electron micrographs, and C. Hubert, E. Ruff, S. Ahmerkamp and N. Dubilier for discussions. This study was supported by European Research Council starting grant MASEM 242635 (M.S., E.H., J.C.), the Campus Alberta Innovation Chair Program (M.S., E.H., X.D.), the Canadian Foundation for Innovation (M.S), the Alberta Small Equipment Grant Program (M.S.), the German Federal State Nordrhein-Westfalen (M.S.), the Max Planck Society, and the Natural Sciences and Engineering Research Council of Canada for a Banting fellowship to M.K. and a Discovery Grant to M.S.
Author information
Authors and Affiliations
Contributions
E.H. and J.C. performed sampling, cultivation and physiological experiments. M.K. performed proteomics and data analysis. D.R. performed transmission electron microscopy. S.L. and E.H. performed scanning electron microscopy. E.H. performed CARD-FISH imaging. H.T. performed next-generation sequencing, H.G.-V. performed read processing, assembly and binning. E.H. performed in silico processing of next-generation sequencing data with assistance from H.G.-V., X.D. and M.S. M.W.B., C.W.S. and A.J.R. analysed sequences for Arcobacter associated with S. tetraspora. E.H., B.V. and K.B performed chemical analysis with input from J.M, K.-U.H. and M.S. The experimental design was developed jointly by M.S., E.H., J.M., M.K. and K.-U.H. E.H. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Micrographs for L. limosa and epibiotic Arcobacter.
a, Scanning electron micrograph showing L. limosa and associated bacteria. Pilus (1) connecting Arcobacter (5) with L. limosa. Pseudopodial extensions (2) are used for the acquisition of prey bacteria (3) (Alteromonas). Short anterior flagellum (4). Long posterior flagellum (6). b–e, CARD-FISH labelling with probes targeting the SSU rRNA of L. limosa (Euk516 in red) and Arcobacter (Epsy914 in green). The scale bar applies to all figures. f–i, CARD-FISH labelling of L. limosa with probes targeting the SSU rRNA of A. macleodii (Alt184 in green). The scale bar applies to all figures. j–r, Transmission electron micrographs showing different structural features of L. limosa. Mitochondria-related organelle (mro), nucleus (nucl), digestive vacuoles (dv), double basal body (bb), endoplasmic reticulum (er), inner (im) and outer membrane (om), tubular cristae (cristae), extracellular matrix (ex), bacterium (bac), membrane (mem), flagellum (flag), multivesicular body (mb). For a–i, each specimen shown represents at least ten specimens for which images were recorded.
Extended Data Figure 2 Relative abundance of L. limosa and co-enriched bacteria under different growth conditions.
The abundance of L. limosa and its associated microbiota was determined at three different conditions (treatments) with three independent experiments per treatment: 1–3, presence of nitrous oxide and prey bacteria; 4–6, absence of nitrous oxide and presence of prey bacteria; 7–9, presence of nitrous oxide, dissolved organic nutrients and hydrogen and absence of prey bacteria. Relative abundances were determined via proteomics and estimated on the basis of the total normalized spectrum count per population.
Extended Data Figure 3 Genome statistics for L. limosa and epibiotic Arcobacter.
The pie chart represents the classifications of gene models into functional categories for Arcobacter. Gene classifications were performed with the RAST functional annotations and the SEED subsystem database32.
Extended Data Figure 4 A new type of NAD(P)H-dependent Fe-hydrogenase.
The genome of L. limosa encoded a so far undescribed NAD(P)H-dependent Fe-hydrogenase. Genes with identical domain architecture were also identified in P. biforma and T. vaginalis (shown in bold type). The scale bars represent substitution rate per site. a, Phylogeny of the Fe-hydrogenases domain. b, Phylogeny of the NAD/NADP binding domain. Phylogenies were inferred by RAxML using the WAG amino-acid replacement matrix. c, Domain architecture of the NAD(P)H-dependent Fe-hydrogenase (2) compared with the domain architecture of Fe-hydrogenase (3) and the NADPH accepting domain of the cyt P450 reductase (1). The scale bar shows approximate amino-acid positions. d, Predicted electron flow within the NAD(P)H-dependent Fe-hydrogenase indicates the capability for a proton-dependent recycling of NAD(P)H. Note: the shape of the model does not intent to depict the actual three-dimensional structure of the protein.
Extended Data Figure 5 Maximum likelihood tree of quinone-reactive Ni/Fe-hydrogenases (subunit hydB).
The tree shows the phylogenetic relation of quinone-reactive Ni/Fe-hydrogenases from Arcobacter associated with S. tetraspora, P. biforma and L. limosa (indicated in red). Circles represent bootstrap support values for each node. The scale bar represents substitution rate per site.
Extended Data Figure 6 The fitness of L. limosa depends on its symbiont.
Syntrophy was enabled by the presence of nitrous oxide acting as electron acceptor for bacterial hydrogen oxidation. a, Inhibition of nitrous oxide reduction (addition of the competitive inhibitor acetylene, see arrow) led to a reduced growth of L. limosa and reduced respiration rates. To monitor respiration rates, 13C-enriched Alteromonas were added together with acetylene. Digestion of 13C-labelled bacteria by L. limosa led to the production of 13C-bicarbonate, which was measured after conversion to 13CO2 (right). Similar effects on the growth and respiration rates were observed after adding hydrogen (b) or hydrogen and acetate (c) to a culture. d, Growth of L. limosa and production of hydrogen and fatty acids while growing syntrophically (nitrous oxide present). e, Growth of L. limosa in the presence of antibacterial antibiotics (nitrous oxide absent). f, Growth of L. limosa in the presence of antibacterial antibiotics (nitrous oxide present). g, Growth of L. limosa in the presence of nitrate (2 mM) and oxygen (0.2 mM). Growth of L. limosa was compared with a culture that contained nitrous oxide (2.2 mM) and with a control culture that did not contain an electron acceptor for hydrogen oxidation. Each panel shows the results of at least five independent experiments, with cell numbers depicted as averages of seven cell counts per experiment; error bars, s.d.
Extended Data Figure 7 Expression levels for Arcobacter proteins involved in attachment and chemotaxis.
a. Expression level of proteins involved in attachment in the presence (red) and absence of L. limosa (blue). b, Expression level of proteins involved in chemotaxis. Expression levels were measured and averaged for three independent experiments per treatment (see also Extended Data Fig. 2). Error bars, s.d. See Supplementary Table 1 for gene accession numbers and statistical tests.
Extended Data Figure 8 Domain architecture of L. limosa fibronectin type III domain-containing proteins.
Protein architectures and conserved protein domains were identified using the SMART protein domain detection tools. See Supplementary Table 1 for gene accession numbers and expression levels.
Supplementary information
Supplementary Table 1
Accession numbers and per gene expression levels as determined with proteomics. (XLS 273 kb)
Supplementary Table 2
Calculation of the thermodynamic and kinetic feasibility of hydrogen transfer between L. limosa and Arcobacter. (XLS 10 kb)
Supplementary Information
This file contains Supplementary Notes regarding the diagnosis of Lenisia limosa gen. et sp. Nov (PDF 80 kb)
Rights and permissions
About this article

Cite this article
Hamann, E., Gruber-Vodicka, H., Kleiner, M. et al. Environmental Breviatea harbour mutualistic Arcobacter epibionts. Nature 534, 254–258 (2016). https://doi.org/10.1038/nature18297
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18297
This article is cited by
-
Phagotrophic protists preserve antibiotic-resistant opportunistic human pathogens in the vegetable phyllosphere
ISME Communications (2023)
-
The microbiome of a bacterivorous marine choanoflagellate contains a resource-demanding obligate bacterial associate
Nature Microbiology (2022)
-
Emergent “core communities” of microbes, meiofauna and macrofauna at hydrothermal vents
ISME Communications (2021)
-
Horizontal acquisition of a patchwork Calvin cycle by symbiotic and free-living Campylobacterota (formerly Epsilonproteobacteria)
The ISME Journal (2020)
-
Improved culture enrichment broth for isolation of Arcobacter-like species from the marine environment
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
