
Abstract
Implantation of embryonic stem cells (ESC)-derived insulin-producing cells has been extensively investigated for treatment of diabetes in animal models. However, the in vivo behavior and migration of transplanted cells in diabetic models remains unclear. Here we investigated the location and migration of insulin-producing cells labeled with superparamagnetic iron oxide (SPIO) using a dynamic MRI tracking method. SPIO labeled cells showed hypointense signal under the kidney subcapsules of diabetic mice on MRI and faded gradually over the visiting time. However, new hypointense signal appeared in the spleen 1 week after transplantation and became obvious with the time prolongation. Further histological examination proved the immigrated cells were insulin and C-peptide positive cells which were evenly distributed throughout the spleen. These intra-spleen insulin-producing cells maintained their protective effects against hyperglycemia in vivo and these effects were reversed upon spleen removal. Transplantation of insulin-producing cells through spleen acquired an earlier blood glucose control as compared with that through kidney subcapsules. In summary, our data demonstrate that insulin-producing cells transplanted through kidney subcapsules were not located in situ but migrated into spleen and rescues hyperglycemia in diabetic models. MRI may provide a novel tracking method for preclinical cell transplantation therapy of diabetes continuously and non-invasively.
Similar content being viewed by others


Introduction
Type 1 diabetes is characterized by the selective destruction of pancreatic β-cells caused by an autoimmune attack. Type 2 diabetes presents a more complex etiology including β-cell loss caused by apoptotic programs and peripheric insulin resistance. Restoration of damaged β-cells by transplantation from exogenous sources or by endocrine pancreas regeneration would be ideal therapeutic options for diabetes. The success in restoring normoglycemia by islet transplantation indicates that cell replacement therapy of this severe disease is achievable. However, this therapy is not widely used because of the severe shortage of transplantable donor islets1,2,3. Embryonic stem cells (ESCs), which are telomerase-positive, immortal and capable of both self-renewal and differentiation into all cell types of the body4,5, could potentially supply an unlimited number of pancreatic cells for transplantation into diabetic patients.
Many studies have demonstrated that ESC can differentiate into insulin-producing cells and ultimately rescue hyperglycemia in diabetic mice6,7,8,9. However, the in vivo behavior of transplanted insulin-producing cells in diabetic models needs further investigation. Until now, the major means to determine whether stem cell-mediated therapeutic interventions yield significant performance improvements are glucose level and pancreatic function assessments in vivo. Information regarding the location, distribution and migration of transplanted insulin-producing cells in diabetic models has been obtained via histological means, which suffer from significant shortcomings, including the scarification of modeled animals at scheduled time points, a lack of longitudinal observations in the same living organisms and limited utility for clinical studies. Thus, a method for evaluating cell distribution and migration over time in a noninvasive manner is urgently needed for both animal studies and future clinical trials in stem-based studies.
Cell labeling for high-resolution magnetic resonance imaging (MRI) with paramagnetic contrast agents is a well-suited tool providing detailed anatomic information in a noninvasive manner. This technology has been used to characterize histopathology and morphologic phenotypes10,11,12,13. The value of MRI in monitoring and tracking stem cells transplanted into host tissues has been established for heart, kidney and cerebral diseases14,15,16,17. For cellular MRI studies, superparamagnetic iron oxide (SPIO) particles with various advantages were the most commonly used contrast agents for cell labeling22,23,24,25 and areas containing SPIO-labeled cells appear as regions of low signal intensity on MRI images, creating negative contrast. Although a few MRI-related studies have been reported to successfully visualize the location of islets via magnetic nanoparticle imaging in vivo18,19,20,21, however, to date, few reports using MRI visualized the migration of transplanted insulin-producing cells in vivo continuously and dynamically. Furthermore, correlating the migration site shown through MRI, we aim to evaluate and compare the therapeutic efficiencies of transplanted insulin-producing cells via different transplantation sites.
Here, we show that SPIO labeled insulin-producing cells demonstrated hypointense signal under the kidney subcapsules of diabetic mice on MRI but faded gradually over the visiting time. However, new hypointense signal appeared in spleen 1 week after transplantation and persisted until the end of the visiting time, which was further confirmed through histological methods. The final glucose measurement results demonstrated that although the migration of transplanted cells occurred, these intra-spleen insulin-producing cells maintained their protective effects against hyperglycemia in vivo and these effects were reversed upon spleen removal. The study of different transplantation sites showed that transplantation of insulin-producing cells through spleen acquired an earlier blood glucose control as compared with that through kidney subcapsules.
Results
Insulin-producing cells from ESC were efficiently labeled with SPIO
ESCs were induced to differentiate into insulin-producing cells, as reported previously (the differentiation strategy is illustrated in Figure 1A.)26. After the treatment of activin A and sodium butyrate (endoderm cell differentiation), about 20% of cells expressed PDX-1. At the final differentiation stage, Insulin and C-peptide antibody stainings were present in 93% and 89% of the differentiated cells, respectively. In the present study, Prussian blue staining revealed that insulin-producing cells were successfully labeled with SPIO at various concentrations (10, 25, 50 and 100 μg/ml) (Figure 1B). The labeling efficiency was gradually increased and the labeling efficiency reached a maximum when cells were labeled with 50 μg/ml (Figure 1C). Transmission electron microscopy confirmed the cellular uptake of SPIO particles and the presence of SPIO particles inside the cytoplasms of labeled cells (Figure 1D).

SPIO labeling of insulin-producing cells from ESC.
(A) A schematic presentation of the differentiation strategy used in this study. (B) Prussian blue staining shows the uptake of iron particles into insulin-producing cells labeled with different SPIO concentrations (i.e., 0, 10, 25, 50 and 100 μg/ml). Cells labeled with Feridex alone were used as the control. (C) Graphs show labeling efficiency based on the results of four separate experiments. *p < 0.05 vs. 10 μg/ml SPIO treated group. #p < 0.05 vs. 25 μg/ml SPIO treated group. (D) Transmission electron microscopy images of labeled and unlabeled insulin-producing cells. Black arrows indicate SPIO particles in the cytoplasm.
SPIO labeling does not affect cell proliferation and β-cell differentiation capacity
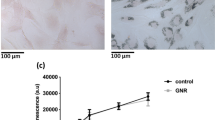
After labeling with different SPIO concentrations for 4 h, more than 95% of insulin-producing cells remained viable. After SPIO particle uptake and further culturing for 24 h (Figure 2 A,B), 48 h and 72 h (Supplementary Figure S1), there were also no significant changes in cell viability or proliferation for the SPIO-labeled insulin-producing cells. Flow cytometry analysis results revealed no significant changes in cell viability among the groups treated with different SPIO concentrations. Furthermore, the expression of pancreatic-specific genes, such as PDX-1, PAX-6, insulin 1, insulin 2 and Glut2, did not reveal any significant differences between labeled and unlabeled cells (Figure 2 C, D). Insulin and C-peptide immunofluorescence also demonstrated that SPIO labeling did not alter protein expression in labeled vs. unlabeled cells (Figure 2 E). To investigate the functional status of labeled and unlabeled cells, we further analyzed glucose-dependent insulin release and determined that there were no differences in basal and glucose-secreted insulin release in labeled vs. unlabeled cells. Insulin release was nearly six-fold higher in the high-glucose medium than in the low-glucose medium, regardless of cell labeling status (Figure 2F).

Effects of SPIO labeling on the proliferative activity and differentiation capacity of labeled insulin-producing cells.
(A) 4 hours after labeling with different concentrations (i.e., 0, 10, 25, 50 and 100 μg/ml) of SPIO, cells were further cultured for 24 h and cell viability was measured by flow cytometry analysis. Graphs show the cell viability results of three independent experiments. (C, D) Pancreatic specific gene expression of labeled and unlabeled insulin-producing cells was analyzed by RT-PCR and real-time PCR. β-actin was served as control to ensure the equal amounts of RNA. Histogram plots represented the real-time PCR results of three independent experiments. (E) At the final differentiation stage, labeled and unlabeled cells were immunostained with antibodies against insulin and C-peptide. The blue color in the images depicts the nucleus stained by DAPI. Scale bar: 50 μm. (F) The amount of insulin released in the supernatants was measured by ELISA and normalized to protein content after cells were incubated with low-glucose (3 mM) or high glucose (22 mM) for 0.5 h. Values were given as means ± SD.
In vitro MRI shows the hypointense signal changes with different cell numbers and SPIO concentrations
Labeled insulin-producing cells manifested as hypointense signal (dark area) on all imaging sequences but were most obvious on T2*-weighted imaging. The signal intensity decreased because of differing SPIO-containing nanoparticle uptake into the cells. With increasing cell numbers and SPIO concentrations, the hypointense signal became more obvious (Figure 3A, C). A significant signal change was noted when cells were labeled with 50 μg/ml SPIO. Thus, 50 μg/ml SPIO was used in the following study. Only slight changes were observed in Feridex-only-labeled cells (Figure 3C, D).

In vitro MRI of labeled and unlabeled cells.
(A) Different numbers (1 × 104, 5 × 104, 1 × 105, 5 × 105, 1 × 106 cells) of SPIO-labeled and unlabeled insulin-producing cells were detected by MRI. Unlabeled cells were used as controls. (B) Graphs show the relative intensity of “dark spots” of different cell numbers of three independent experiments. (C) A total of 5 × 105 cells labeled with different concentrations of SPIO (0, 10, 25, 50, 100 μg/ml) were collected for in vitro MRI detection. Unlabeled cells and cells labeled with Feridex alone were used as the controls. (D) Graph of the relative “dark spot” intensities of different SPIO concentrations, as measured in three independent experiments.
Dynamic in vivo MRI demonstrates the hyposignal changes in kidney and spleen
After transplantation into the kidney subcapsules, labeled cells were detected as well-defined hypointense signals (black area in kidney on MRI images) in all animals, which were consistent with the presence of intra-cellular SPIO (Figure 4A,C). In contrast, the control mice (injected with unlabeled insulin-producing cells) lacked the hypointense signal (Supplementary Figure S2). Pseudo-colorized images from the individual four main panels were shown to better demonstrate the high signal intensity of labeled cells in the affected region (Figure 4B, D). Dynamic MRI showed the signal intensity in kidney decreased gradually after transplantation. The MRI signal intensity under kidney subcapsules decreased nearly 60% 6 weeks after the cell delivery (Figure 4E). Notably, new hypointense signals appeared in the spleen 1 week after injection into kidney subcapsules, became more visible and persisted until the end of the observation time. A 2.5-fold higher MRI signal intensity in spleen was found at the end of the visiting time as compared with that of the beginning (Figure 4F). As for the animals transplanted with boiled labeled cells (used as control), hypointense signals (black areas) could be visualized at the moment of transplantation and were still visible 1 week after injection; afterwards, these hypointense signals disappeared gradually. No hypointense signal could be detected in spleen and other organs after transplantation of boiled labeled cells (Supplementary Figure S2).

In vivo MRI of cell transplantation into kidney subcapsules over a six week period.
SPIO-labeled insulin-producing cells were transplanted into the renal subcapsular area. Abdominal MRI was performed before and 1, 2, 4 and 6 weeks after transplantation. White arrows indicate the hypointense signal at the region of cell transplantation (A) and in the spleen (C). The color images (B, D) are pseudo-colorized images from the individual four main panels to better demonstrate the high signal intensity of labeled cells in the affected region of interest using ImageJ software. The blue area in spleen indicated the hypointense signal change on T2*-weighted imaging. (E, F)Histogram indicated the relative intensities of MRI signals in kidney (E) and spleen(F) over time. Values of hypointense signal areas were corrected with the intensities of the psoas major muscles.*p < 0.05 versus MRI intensities of hypointense signal areas at the beginning.
Live insulin-producing cells migrate from the kidney subcapsules into the spleen
Prussian blue staining results at the end of the observation time demonstrated that only small numbers of Prussian blue stain-positive particles could be found under the kidney subcapsules and numerous Prussian blue stain-positive cells were distributed diffusely in the spleen (Figure 5A, Supplementary Figure S3, S4). To exclude the possibility that the Prussian blue stain-positive particles in the spleen might arise from resident macrophages that engulfed the iron-labeled nanoparticles, further histological studies of insulin and C-peptide expression in the spleen, kidney and liver (Figure 5B, Supplementary Figure S3) were also performed. The results demonstrated that insulin- and C-peptide-positive cells could be measured in the hypointense signal area and demonstrated a distribution manner similar to the Prussian blue stain-positive particles in the left kidney and spleen, which suggested that the “black spots” in the spleen were live insulin-producing cells that migrated from the kidney subcapsules rather than macrophages that took up SPIO particles. No insulin or C-peptide-positive cells could be identified in the liver, which was consistent with the results of the liver MRI imaging (Figure 5B).

Histology of transplanted cells.
(A) 6 weeks after cell transplantation, the spleen, kidney and liver tissues were analyzed for Prussian blue staining (A, original magnification × 100). Original magnification of the right image of each tissue was 200×.The blue particles indicate Prussian blue-positive sites. (B) Immunofluorescence staining for insulin and C-peptide was performed in all tissues (original magnification × 100). The green and red colors depict insulin (FITC -conjugated) and C-peptide protein. The blue color in the images depicts the nucleus, stained by DAPI. The merged images by insulin/C-peptide and nucleus were obtained after superposition of the green and blue channels. Scale bar: 100 μm.
Intra-spleen migration of ESC-derived insulin-producing cells rescues hyperglycemia in diabetic mice
The cell-transplanted mouse blood glucose levels (n = 20), including the levels in mice transplanted with labeled (n = 10) and unlabeled cells (n = 10), gradually decreased 8 days post-transplantation into kidney subcapsules (Figure 6A). At the end of the observation time, the average blood glucose levels of cell-transplantation group, regardless of labeling (Figure 6A), decreased to 10.79 ± 1.92 mM, which was lower than that of the control group (PBS injection).In order to verify the function of migrated insulin-producing cells in spleen, the blood glucose-rescued mice(n = 10) underwent a splenectomy 4 weeks after transplantation and the results demonstrated that the mice again became hyperglycemic within 3 days after the operation compared with the sham operation mice and hyperglycemia was maintained for at least 2 weeks (Figure 6A). In the sham operation group, all the mice exhibited stable euglycemia. At the same time, we also performed splenectomies in PBS-injection group and found the blood glucose in PBS-injection group remained stable hyperglycemia even after spleen removal. The survival rates of kidney transplantation group were 62.5%, much higher than the survival rate in the control group (37.5%) (Figure 6B).

Blood glucose levels and survival rates of the transplanted cell and control groups.
(A) Blood glucose was measured every 4 days in the labeled(red lines), unlabeled cells(blue lines) and control groups(green lines) over a 4 week period. 4 weeks after transplantation, splenectomy was performed in blood glucose-rescued mice (a) and PBS-injection group(c). A sham operation was also performed in either blood glucose-rescued mice (b) or PBS-injection group (d). Blood glucose was measured every 7 days after operation. (B) The survival rates of the transplanted cell (red lines) group and control group (blue lines) were compared. (C) Blood glucose was measured in the groups transplanted into kidney subcapsules (blue lines) or into spleen (red lines). (D) The survival rates of kidney transplantation group (green line) and spleen transplantation group (red line) were calculated.
Correlating the migration site shown through MRI, we also compared the glucose levels and survival rates of cell-transplantation into kidney subcapsules to those of cell transplantation into spleen. Results demonstrated that normoglycemia was reached day 4 post-transplantation into spleen, earlier than that of kidney subcapsules transplantation (Figure 6C). At the final observation time, the survival rates of kidney and spleen transplantation group were both 62.5%, without statistical significance (Figure 6D).
Discussion
Using dynamic and non-invasive MRI tracking method, we demonstrated SPIO labeled insulin-producing cells were not located in the injection area but migrated to the spleen over the entire 42-day study period,which was further proved through histological examination. Despite intra-spleen cell migration, these transplanted insulin-producing cells maintained their protective effect against hyperglycemia in vivo. Transplantation of insulin-producing cells through spleen acquired an earlier blood glucose control as compared with that through kidney subcapsules.
Many studies have demonstrated that ESC-derived insulin-producing cells could rescue diabetes in mice27,28,29,30,31. However, the research about the location, distribution and migration of transplanted insulin-producing cells in diabetic models required longitudinal and dynamic observations for quantifying graft survival and migration. The capacity to label stem cells with magnetic resonance contrast medium has created the potential for the longitudinal, noninvasive tracking of transplanted cells. SPIO is an FDA-approved MRI contrast agent with high T2 relativity that has been used extensively to improve diagnostic imaging32. After SPIO labeling of ESC-derived insulin-producing cells, no significant decreases were found in cell viability. However, the effect of SPIO labeling on cell differentiation capacity remains controversial33,34,35. Our results demonstrated that SPIO labeling did not have any observable effects on pancreatic differentiation potential of ESC and did not affect differentiated insulin-producing cell gene and protein expression. In a previous study, the incubation of islets in medium containing Resovist (clinically approved SPIO, Bayer Schering Pharma AG) for 48 h diminished insulin production36,37,38, while Kim's research demonstrated that islets remained functional after SPIO labeling, as determined with in vitro viability and insulin secretion tests39. Our glucose-stimulated insulin secretion (GSIS) test proved that SPIO labeling did not affect the insulin secretion function in differentiated insulin-producing cells.
Using MRI, we found that labeled cells showed hypointense signal (dark areas) at the injection site on the beginning and these hypointense areas were still present but weakened at the injection sites 6 weeks after transplantation. The presence of iron nanoparticles in tissue is evident primarily because of a darkening effect on T2- or T2*-weighted MR images. Remarkably, the T2* effect of SPIO particles resulted in hypointense signals that obviously exceeded the labeled stem cell mass, which, in essence, constituted a powerful signal amplification tool. It was once reported that SPIO-based imaging was not well suited for long-term stem cell monitoring39,40,41 because SPIO may not stay in the transplanted cells over time but instead may be phagocytosed by macrophages and other cell types, resulting in an uncoupling between the magnetic resonance signal and stem cell viability42. However, isolated SPIO nanoparticle-labeled mouse islets can be effectively and safely imaged using MRI up to 18 weeks after transplantation43. Our results demonstrated that labeled ESC-derived insulin-producing cells could indeed survive up to 6 weeks after transplantation.
In our present research, cell migration was observed through dynamic MRI. Results revealed that most of the labeled cells were not located in the graft site, but migrated to the spleen with time, which has not been reported previously. On MRI, hypo-intense signals appeared in the spleen 1 week after transplantation, became more obvious at the 4 week and persisted over the entire visiting time. Further histological confirmation proved that hypo-signal areas in the spleen were positive for Prussian blue staining and, more importantly, were positive for insulin and C-peptide expression. These findings indicated that the MRI hypo-signal correlated with graft survival, that is to say, the labeled insulin-producing cells not resident macrophages that engulfed the iron nanoparticles migrated into the spleen from the kidney subcapsules, causing hypointense signals in the spleen. These Prussian blue stain-positive particles were distributed diffusely in the spleen. From the distribution from of transplanted cells, we speculate the insulin-producing cells migrate from kidney into spleen through blood circulation system, not from kidney to spleen directly. However, the exact mechanisms involved in grafted cell migration remain uncertain.
Despite cell migration, these transplanted insulin-producing cells partially rescued diabetes in the recipients. Both glucose tests and survival rate analysis demonstrated statistically significant improvements in the cell-treated animals at the end of the visiting time. No such functional improvement was observed in the control group (PBS injection). This finding is in agreement with previous reports suggesting that ESC-derived insulin-producing cells normalize blood glucose levels and increase survival rates in diabetic mice (8,9,44). To further verify the function of migrated insulin-producing cells in spleen, some mice with normal glycemia after cell transplantation underwent splenectomies. The glucose-rescued mice regained hyperglycemia as compared with sham-operation group, which further verified the function of migrated insulin-producing cells into the spleen.
Correlating the migration site shown through MRI, we then compared the therapeutic efficiencies of transplanted insulin-producing cells via different transplantation sites. Our study of cell transplantation into the renal subcapsular space has suggested a delayed function where normoglycemia was not reached until day 8 posttransplantation. However, normoglycemia was reached day 4 posttransplantation into spleen, earlier than that of kidney subcapsules transplantation. Kidney subcapsules has been proposed as a suitable location for the transplantation of islet and stem cell-derived insulin-producing cells in many rodent models44,45,46,47. It is relatively easy to use and results in the reversal of diabetes within days. Unfortunately, the kidney subcapsules has a relatively poor blood supply and does not provide an oxygen-rich microenvironment for the islets. Theoretically, insulin producing cells transplanted into the spleen would be exposed to a similar environment as that of their native pancreas, with a good vascular supply and physiological delivery of insulin into the splenic vein. In fact, the spleen is a metabolically suitable site for insulin producing cells transplantation. Our results demonstrated that transplantation of insulin-producing cells through spleen acquired an earlier blood glucose control as compared with that through kidney subcapsules. However, the survival rates of kidney and spleen transplantation group showed no statistical significance.
In conclusion, for the first time, our study demonstrated that insulin-producing cells under the kidney subcapsules were not located in situ but migrated into spleen and also rescues hyperglycemia in diabetic mice. Transplantation of insulin-producing cells through spleen acquired an earlier blood glucose control as compared with that through kidney subcapsules. As a noninvasive method, MRI permits longitudinal studies on the same animal, which would increase our understanding of stem cell behavior after transplantation and could aid in the development of effective and translational stem cell-based diabetes therapies.
Methods
Ethics statement
All animal experiments were performed in accordance with the guidelines of China Council on Animal Care and Use. All animal procedures carried out in this study were reviewed and approved by the Institutional Animal Care and Use Committee of the Ethics Committee of Sun Yat-sen University of Medicine, China. All animals were kept and the experiments were performed in accordance with the European Community guidelines for the use of experimental animals (86/609/EEC).
Cell culture and in vitro pancreatic differentiation
ESCs were induced to differentiate into insulin-producing cells as previously described26. Undifferentiated E14 mouse ES cells (ATCC, Manassas, VA) were suspended in the differentiation medium and were cultured to develop into embryoid bodies (EBs) for 4 days. The EBs were then cultured in the differentiation medium containing 25 ng/ml activin A (Sigma–Aldrich) for 48 h and then switched to the differentiation medium supplemented with 1 mM sodium butyrate. After 3 days of sodium butyrate treatment, the differentiated cells were further matured in medium containing 10 ng/ml basic fibroblast growth factor and 10 ng/ml hepatocyte growth factor for 7 days, followed by 10 mM nicotinamide (Sigma–Aldrich) for another 7 days.
Cell labeling, labeling efficiency and cell viability
Differentiated insulin-producing cells were labeled using SPIO (Feridex; Berlex, Wayne, NJ) as a contrast agent and protamine sulfate (Sigma-Aldrich, St. Louis, MO) as a transfection agent. Cells labeled with Feridex alone were used as the control. For labeling, 1 × 105 cells were incubated in serum-free culture medium containing SPIO for 4 h. SPIO concentrations were 10, 25, 50 and 100 μg/ml and the final protamine sulfate concentration was 16 μg/ml. After 4 h of incubation, cells were washed twice with PBS to remove excess contrast agents. Intracellular SPIO was detected via transmission electron microscopy. The protocols were described in Supplementary Materials and Methods. Percent labeling efficiency was assessed by Prussian blue staining (at least 200 cells were counted in five randomly chosen microscopic fields). For cell viability, 1 × 105 cells were exposed to different SPIO concentrations (10, 25, 50 and 100 μg/ml) for 4 h. Furthermore, to study the effect of intracellular SPIO particles on cell proliferation, labeled cells were further cultured for different times (24, 48 and 72 h). Flow cytometric analysis was used to measure cell viability. The protocols of Prussian blue staining and flow cytometric analysis were described in Supplementary Materials and Methods.
Reverse transcription PCR (RT-PCR), real-time quantitative PCR and Immunofluorescence
RT-PCR and real-time PCR were performed to measure the mRNA expression levels of pancreatic markers such as PDX-1, PAX-6, Glut2, insulin 1 and insulin 2 in labeled and unlabeled cells. Primer sequences are shown in Supplementary Table 1. The insulinoma cell line NIT-1 was used as a positive control. Insulin and C-peptide expression were examined via immunofluorescence. Rabbit anti-mouse insulin and C-peptide primary antibodies (1:100; Cell Signaling Technology, MA, USA) and appropriate fluorescein isothiocyanate (FITC)-conjugated and Cy3-conjugated secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) were used. Nuclear DNA was dyed with DAPI (Sigma-Aldrich, MO, USA). Images were captured using an Olympus IX-71 microscope and a LEICA microscope.
Insulin secretion assessment by enzyme-linked immunosorbent assay (ELISA)
Labeled or unlabeled insulin-producing cells (5 × 105) were washed and preincubated in Krebs-Ringer bicarbonate buffer (KRB) containing 3 mM glucose. After preincubation for 30 minutes, the buffer was replaced with fresh KRB containing 3 or 20 mM glucose. After 30 minutes of incubation, medium was collected and the medium insulin content was measured using an insulin ELISA kit (Millipore, MA, USA). The intracellular protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Bio-Rad, CA, USA). Medium insulin levels (insulin secretion) were normalized based on the respective intracellular protein content.
In vitro MRI
In vitro MRI was performed using a 1.5-T MR scanner (Philips Medical Systems, Best, the Netherlands) with an 11-cm circular surface coil. To determine the optimal labeling concentration, 5 × 104 cells labeled with different SPIO concentrations (0, 10, 25, 50 and 100 μg/ml) were collected. To determine the minimal detectable number of insulin-producing cells, different numbers (1 × 104, 5 × 104, 1 × 105, 5 × 105 and 1 × 106 cells) of labeled cells (under optimal labeling concentrations) and unlabeled cells were collected. The cells were then washed and centrifuged and the centrifuged cell pellets were imaged within the test tubes. The tubes were placed in a plastic box filled with 37°C water to ensure signal homogeneity and to avoid potential susceptibility artifacts from the surrounding air. Each subset of imaged cells contained labeled cells and unlabeled control cells.
Animal model and cell transplantation
Diabetes was induced in nude mice with a single intraperitoneal streptozotocin injection (180 mg/kg). Mice with blood glucose levels above 13.9 mM for 7 consecutive days were considered to be diabetic. 5 × 106 labeled or unlabeled differentiated insulin-producing cells were transplanted into the kidney subcapsules (20 mice).The same volume of PBS without differentiated cells was injected into other 10 diabetic mice as a control. In some experiments, 5 × 106 labeled insulin-producing cells were transplanted into the kidney subcapsules of 10 mice and the same numbers of labeled insulin-producing cells were transplanted into spleen of another 10 diabetic mice. Additionally, dead (incubated in boiling water for 10 minutes) labeled cells were transplanted into another 3 animals to confirm the label retention in the viable cells.
In vivo MRI
To assess the distribution and migration of labeled insulin-producing cells in vivo, abdominal MRI was performed before injection and at 1, 2, 4 and 6 weeks after cell injection. MRI was performed using a 1.5-T MR scanner (Philips Medical Systems, Best, the Netherlands) with a 5-cm linearly polarized birdcage radio frequency mouse coil (Shanghai Chen Guang Medical Technologies Co. LTD, China). Coronal and transverse fast field echo T2*-weighted images were acquired using the following parameters: TR/TE, 200/14.7 ms; flip angle, 25°; FOV, 80 mm; matrix, 256 × 256; section thickness, 1 mm; section gap, 0 mm. The presence and migration of labeled cells in vivo were assessed and compared with unlabeled cells and PBS. MRI sequences were shown in Supplementary materials and methods. DICOM software was used to analyze the relative intensities of MRI signals. Values were corrected with the intensities of the psoas major muscles.
Glucose and survival rate test
Blood glucose was measured every 4 days in the cell-transplanted (transplanted into kidney subcapsules) and control groups with an ACCU-CHEK Advantage Meter glucometer (Roche Diagnostics GmbH, Mannheim, Germany). At the end of a 4-week follow up, the mice whose hyperglycemia was rescued after cell transplantation underwent splenectomies and blood glucose was measured for another 2 weeks. The sham-operated mice in blood glucose-rescued group and those underwent splenectomies in PBS-injection group were both used as control. The diabetic mouse survival rates were also recorded and calculated. In some experiments, the glucose levels and survival rates of cell-transplantation into kidney subcapsules or spleen were evaluated and compared. All of the animal experimental procedures were approved by the Sun Yat-sen University Institutional Animal Care and Use Committee.
Histology analysis
The kidneys, spleen and liver were excised, embedded, frozen and cut into 5-μm-thick slices 6 weeks after cell transplantation. Prussian blue staining was performed to visualize the iron-positive cells within these tissues. To determine the in vivo pancreatic differentiation capacity, insulin and C-peptide expression was detected using immunofluorescence. Rabbit anti-mouse insulin and C-peptide primary antibodies (1:100; Cell Signaling Technology, MA, USA) were used with corresponding FITC-conjugated and Cy3-conjugated secondary antibodies (Sigma).
Statistical analysis
All of the in vitro experiments were repeated at least three times. The data were expressed as means ± standard deviations and were analyzed using the SPSS 14.0 software. ANOVA was used to identify significant differences and p < 0.05 was considered to be statistically significant.
References
Roche, E. et al. Insulin-secreting cells derived from stem cells: clinical perspectives, hypes and hopes. Transpl Immunol 15, 113 (2005).
Ryan, E. A. et al. Five-year follow-up after clinical islet transplantation. Diabetes 54, 2060 (2005).
Roche, E., Santana, A., Vicente-Salar, N. & Reig, J. A. From stem cells to insulin-producing cells: towards a bioartificial endocrine pancreas. Panminerva Med 47, 39 (2005).
Smith, A. G. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol 17, 435 (2001).
Kubo, A. et al. Development of definitiveendoderm from embryonic stem cells in culture. Development (Cambridge, England) 131, 1651 (2004).
Brolen, G. K., Heins, N., Edsbagge, J. & Semb, H. Signals from the embryonic mouse pancreas induce differentiation of human embryonic stem cells into insulin-producing beta-cell-like cells. Diabetes 54, 2867–2874 (2005).
Chen, C., Zhang, Y., Sheng, X., Huang, C. & Zang, Y. Q. Differentiation of embryonic stem cells towards pancreatic progenitor cells and their transplantation into streptozotocin-induced diabetic mice. Cell Biol Int 32, 456–461 (2008).
Jiang, W. et al. In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell Res 17, 333–344 (2007).
Soria, B. et al. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 49, 157–162 (2000).
Gareau, P. J., Wymore, A. C., Cofer, G. P. & Johnson, G. A. Imaging inflammation: direct visualization of perivascular cuffing in EAE by magnetic resonance microscopy. J Magn Reson Imaging 16, 28–36 (2002).
Johnson, G. A. et al. Magnetic resonance histology for morphologic phenotyping. J Magn Reson Imaging 16, 423–429 (2002).
Johnson, G. A., Cofer, G. P., Gewalt, S. L. & Hedlund, L. W. Morphologic phenotyping with MR microscopy: the visible mouse. Radiology 222, 789–793 (2002).
Moore, A. et al. MRI of insulitis in autoimmune diabetes. Magn Reson Med 47, 751–758 (2002).
Himes, N. et al. In vivo MRI of embryonic stem cells in a mouse model of myocardial infarction. Magn Reson Med 52, 1214–1219 (2004).
Ebert, S. N. et al. Noninvasive tracking of cardiac embryonic stem cells in vivo using magnetic resonance imaging techniques. Stem cells (Dayton, Ohio) 25, 2936–2944 (2007).
Daadi, M. M. et al. Molecular and magnetic resonance imaging of human embryonic stem cell-derived neural stem cell grafts in ischemic rat brain. Mol Ther 17, 1282–1291 (2009).
Jung, S. I. et al. In vivo MR imaging of magnetically labeled mesenchymal stem cells in a rat model of renal ischemia. Korean J Radiol 10, 277–284 (2009).
Auer, V. J. et al. Non-invasive imaging of ferucarbotran labeled INS-1E cells and rodent islets in vitro and in transplanted diabetic rats. Curr Pharm Biotechnol 12, 488–496 (2011).
Evgenov, N. V., Medarova, Z., Dai, G., Bonner-Weir, S. & Moore, A. In vivo imaging of islet transplantation. Nat Med 12, 144–148 (2006).
Evgenov, N. V. et al. In vivo imaging of immune rejection in transplanted pancreatic islets. Diabetes 55, 2419–2428 (2006).
Tai, J. H. et al. Imaging islets labeled with magnetic nanoparticles at 1.5 Tesla. Diabetes 55, 2931–2938 (2006).
Lewin, M. et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol 18, 410–414 (2000).
Moore, A., Marecos, E., Bogdanov, A., Jr & Weissleder, R. Tumoral distribution of long-circulating dextran-coated iron oxide nanoparticles in a rodent model. Radiology 214, 568–574 (2000).
Dodd, C. H. et al. Normal T-cell response and in vivo magnetic resonance imaging of T cells loaded with HIV transactivator-peptide-derived superparamagnetic nanoparticles. J Immunol Methods 256, 89–105 (2001).
Moore, A., Josephson, L., Bhorade, R. M., Basilion, J. P. & Weissleder, R. Human transferrin receptor gene as a marker gene for MR imaging. Radiology 221, 244–250 (2001).
Ren, M. et al. Effects of sodium butyrate on the differentiation of pancreatic and hepatic progenitor cells from mouse embryonic stem cells. J Cell Biochem 109, 236–244 (2010).
Bose, B., Shenoy, S. P., Konda, S. & Wangikar, P. Human embryonic stem cell differentiation into insulin secreting beta-cells for diabetes. Cell Biol Int 36, 1013–1020 (2012).
Bruin, J. E. et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res 12, 194–208 (2014).
Gage, B. K., Webber, T. D. & Kieffer, T. J. Initial cell seeding density influences pancreatic endocrine development during in vitro differentiation of human embryonic stem cells. PloS one 8, e82076 (2013).
Kaitsuka, T. et al. Generation of functional insulin-producing cells from mouse embryonic stem cells through 804G cell-derived extracellular matrix and protein transduction of transcription factors. Stem Cells Transl Med 3, 114–127 (2014).
Raikwar, S. P. & Zavazava, N. PDX1-engineered embryonic stem cell-derived insulin producing cells regulate hyperglycemia in diabetic mice. Transplant Res 1, 19 (2012).
Castaneda, R. T., Khurana, A., Khan, R. & Daldrup-Link, H. E. Labeling stem cells with ferumoxytol, an FDA-approved iron oxide nanoparticle. J Vis Exp 57 e3482 (2011).
Farrell, E. et al. Effects of iron oxide incorporation for long term cell tracking on MSC differentiation in vitro and in vivo. Biochem Biophys Res Commun 369, 1076–1081 (2008).
Kostura, L., Kraitchman, D. L., Mackay, A. M., Pittenger, M. F. & Bulte, J. W. Feridex labeling of mesenchymal stem cells inhibits chondrogenesis but not adipogenesis or osteogenesis. NMR Biomed 17, 513–517 (2004).
Au, K. W. et al. Effects of iron oxide nanoparticles on cardiac differentiation of embryonic stem cells. Biochem Biophys Res Commun 379, 898–903 (2009).
Jirak, D. et al. MRI of transplanted pancreatic islets. Magn Reson Med 52, 1228–1233 (2004).
Krejci, J., Pachernik, J., Hampl, A. & Dvorak, P. In vitro labelling of mouse embryonic stem cells with SPIO nanoparticles. Gen Physiol Biophys 27, 164–173 (2008).
Berkova, Z. et al. Vitality of pancreatic islets labeled for magnetic resonance imaging with iron particles. Transplant Proc 37, 3496–3498 (2005).
Kim, H. S., Kim, H., Park, K. S. & Moon, W. K. Evaluation of porcine pancreatic islets transplanted in the kidney capsules of diabetic mice using a clinically approved superparamagnetic iron oxide (SPIO) and a 1.5T MR scanner. Korean J Radiol 11, 673–682 (2010).
Bengel, F. M., Schachinger, V. & Dimmeler, S. Cell-based therapies and imaging in cardiology. Eur J Nucl Med Mol Imaging 32 Suppl 2S404–416 (2005).
Li, Z. et al. Comparison of reporter gene and iron particle labeling for tracking fate of human embryonic stem cells and differentiated endothelial cells in living subjects. Stem cells (Dayton, Ohio) 26, 864–873 (2008).
Chen, I. Y. et al. Comparison of optical bioluminescence reporter gene and superparamagnetic iron oxide MR contrast agent as cell markers for noninvasive imaging of cardiac cell transplantation. Mol Imaging Biol 11, 178–187 (2009).
Juang, J. H. et al. Magnetic resonance imaging of transplanted mouse islets labeled with chitosan-coated superparamagnetic iron oxide nanoparticles. Transplant Proc 42, 2104–2108 (2010).
Kroon, E. et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 26, 443–452 (2008).
Bruin, J. E. et al. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia 56, 1987–1998 (2013).
Santamaria, X., Massasa, E. E., Feng, Y., Wolff, E. & Taylor, H. S. Derivation of insulin producing cells from human endometrial stromal stem cells and use in the treatment of murine diabetes. Mol Ther 19, 2065–2071 (2011).
Hu, Y. H. et al. A secretory function of human insulin-producing cells in vivo. Hepatobiliary Pancreat Dis Int 8, 255–260 (2009).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81370910, 81371607), Research Fund for the Doctoral Program and for the new teacher Program of Higher Education of China (20100171120080), the Natural Science Foundation of Guangdong Province (S2013010016443) and Yat-Sen Scholarship for Young Scientist.
Author information
Authors and Affiliations
Contributions
M.R. and C.S. researched data, performed experiments and wrote the manuscript. X.Z. and R.G. performed MRI examination. G.L. and X.W. researched data. H.C. and J.M. contributed to edit the manuscript. L.Y. and J.S. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. No potential conflicts of interest relevant to this article were reported.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ren, M., Shang, C., Zhong, X. et al. Insulin-producing cells from embryonic stem cells rescues hyperglycemia via intra-spleen migration. Sci Rep 4, 7586 (2014). https://doi.org/10.1038/srep07586
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07586
This article is cited by
-
Challenges with Cell-based Therapies for Type 1 Diabetes Mellitus
Stem Cell Reviews and Reports (2023)
-
MRI tracking of autologous pancreatic progenitor-derived insulin-producing cells in monkeys
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
