
Abstract
Oligonucleotide synthesis is vital for molecular experiments. Bioinformatics has been employed to create various algorithmic tools for the in vitro synthesis of nucleotides. The main approach to synthesizing long-chain DNA molecules involves linking short-chain oligonucleotides through ligase chain reaction (LCR) and polymerase chain reaction (PCR). Short-chain DNA molecules have low mutation rates, while LCR requires complementary interfaces at both ends of the two nucleic acid molecules or may alter the conformation of the nucleotide chain, leading to termination of amplification. Therefore, molecular melting temperature, length, and specificity must be considered during experimental design. POSoligo is a specialized offline tool for nucleotide fragment synthesis. It optimizes the oligonucleotide length and specificity based on input single-stranded DNA, producing multiple contiguous long strands (COS) and short patch strands (POS) with complementary ends. This process ensures free 5′- and 3′-ends during oligonucleotide synthesis, preventing secondary structure formation and ensuring specific binding between COS and POS without relying on stabilizing the complementary strands based on Tm values. POSoligo was used to synthesize the linear RBD sequence of SARS-CoV-2 using only one DNA strand, several POSs for LCR ligation, and two pairs of primers for PCR amplification in a time- and cost-effective manner.
Similar content being viewed by others

Introduction
Long-stranded DNA can be synthesized through DNA synthesis and assembly1. DNA synthesis encompasses chemical synthesis of sequences2, phosphodiesterase synthesis3,4,5 of sequences, and photolithographic synthesis6,7,8,9 using photolithographic techniques and photosensitive vectors to synthesize DNA through UV irradiation and masking. These methods may not be universally available, and direct synthesis techniques face challenges in synthesizing large sequences. The primary challenge involves introducing sequence errors during product synthesis, with error probability positively correlated with sequence length10,11,12. Consequently, sequencing a substantial number of cloned sequences is necessary to minimize error incidence, a cost mitigated by advances in sequencing technology, although the risk of errors remains despite error correction and sequence validation efforts12,13,14,15,16. To further diminish error risk, DNA assembly16,17,18,19,20,21,22,23 offers advantages in long-strand DNA synthesis. By merging multiple small DNA fragments into longer sequences via polymerase chain reaction (PCR) and ligase reaction (LCR), this method proves efficient in terms of accuracy, cost, and time1. Various assembly techniques and their associated software have been published.
Hoover and Lutkovski developed DNAWorks, an automated method for designing and optimizing oligonucleotides for PCR-based gene synthesis24. The software accepts DNA or protein sequences as input and designs optimized oligonucleotides to match the codon bias of the chosen host for expression. Gibson et al. designed the Gibson Assembly by creating sequences complementary at both ends and utilizing T5 exonuclease to excise the complementary sequence at the 5′ end, ensuring that the two assembled fragments produce identical sticky ends22. The Golden Gate assembly, devised and developed by Carola Engler et al. utilizes IIS-type restriction endonucleases to recognize specific sequences of the target gene and create sticky ends, with ligase attaching multiple fragments to the vector plasmid21,25. Jean-Marie Rouillard et al. designed an online tool optimizing the synthesis of long-stranded DNA assemblies through various algorithms26. Currently, oligonucleotide design for LCR or PCR-synthesized genes relies on two parameters: ensuring similar thermodynamic properties (i.e. melting temperature) to ensure uniform hybridization during assembly and high specificity of oligonucleotides for the target to avoid incorrect assembly.
Our POSoligo software was based on the POS method27, utilizing single-stranded DNA as a template, thereby circumventing the need for specific thermodynamic properties in each segment of double-stranded DNA. Moreover, the short patch chains exhibit high specificity for adjacent template chains, thereby reducing the likelihood of mutations during synthesis. In terms of versatility, this software supports the input of single-stranded DNA and RNA sequences.
Materials and methods
All oligonucleotides were procured from Sangon Biotech Bioengineering (Shanghai) Co. A comprehensive index of sequences was compiled by replicating the designs outlined in the procedural section.
Phosphorylation of oligonucleotides
Oligonucleotides (0.1 nmol) underwent phosphorylation at the 5′-end in a PCR tube. The reaction, comprising 3 μL of 10 × PNK buffer (0.5 M Tris/HCl pH 7.6, 0.1 M MgCl2, 50 mM DTE), 2 μL of T4 polynucleotide kinase (10 units), 1 μL ATP (1 mM), and 23 μL nuclease-free H2O, was conducted at 37 ℃ for 30 min. Subsequently, 70 μL of nuclease-free H2O was added, and the reaction was halted by incubating on ice.
Ligase chain reaction
LCR was conducted to covalently link two adjacent long structured oligonucleotides (COS), thus forming full-length sequences. The LCR comprised 1 μL of phosphorylation reaction product, 2.5 μL of 10 × Taq ligase buffer (0.2 M Tris/HCl pH 7.6, 0.25 M potassium acetate, 0.1 M magnesium acetate, 10 mM NAD + , 10% Triton X-100), 1 μL Taq ligase (10 units), and 19.5 μL nuclease-free H2O in a PCR tube. The reaction was carried out on a MyGene series Peltier thermal cycler MG25 + (LongGene, Hangzhou, China) as follows: 95 ℃ for 5 min, 45 cycles of 95 ℃ for 30 s, 51 ℃ for 20 s, and 45 ℃ for 4 s, with a final incubation at 45 ℃ for 5 h.
Polymerase chain reaction amplification
A partial double-stranded DNA template was obtained by PCR amplification using the outermost primer. The reaction mixture comprised 1 μL of LCR product, 2.5 μL of dNTPs (2 mM), 1 μL of each primer (0.2 μM), 5 μL of 10 × Pfu DNA polymerase buffer, high-fidelity DNA polymerase Pfu (5 U/μL, Bio-Basic Inc., Ontario, Canada), and 38.5 μL of nuclease-free H2O. The PCR reaction proceeded as follows: 94 °C for 3 min, 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 2 min, followed by a final extension at 72 °C for 5 min.
Amplification, cloning and sequencing of synthetic fragments
A 6-well plate was prepared with 3 wells of 293FT cells (including duplicate wells and control), at a density of 1 × 106/mL per well. For electro-transfection, 2 μg of gel-recovered DNA and 100 μL of electro-transfection buffer were added to the electro-transfection cup, mixed well in the X-Porator H1 electro-transfer apparatus, and incubated in the incubator after transfection. RNA was extracted from 3 tubes of cytosol (C1, C2, CON) and reverse transcribed to cDNA. The cDNA served as a template, and RBD-F and RBD-R primers (Table 1) were utilized to amplify the RBD target fragment. The reaction proceeded as follows: 94 °C for 3 min, 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 2 min, followed by a final extension at 72 °C for 5 min. The DNA clone was ligated into pGEM-T vector, and 2 μL of the ligation product was transformed into JM109. Recombinant plasmids were screened using the blue/white spot selection method, and the recombinant plasmid was extracted from the white colonies and sequenced for analysis.
Algorithm
Our software employs a sophisticated algorithm to convert the input sequence into oligonucleotides. This input sequence is interpreted as single-stranded DNA, which is then divided into consecutive short strands of 50–120 bps.
Additionally, an auxiliary complementary patch strand is calculated and designed to link the two terminal points of COS, serving as a nexus or bridge. Notably, the two terminal regions of the original sequence intentionally lack patch strands, preserving their accessibility.
Following LCR, primers are carefully crafted in the terminal region of the long chain to facilitate PCR amplification.
Implementation
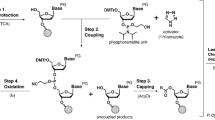
POSoligo has been developed using C + + programming and can be accessed either via direct input sequences or through a .TXT file within the software (Fig. 1). The algorithmic process for POS primarily involves designing a series of overlapping patches, aligning them based on their common sequences, and then merging them to generate the final DNA sequence. This iterative process ensures a satisfactory outcome. The C + + programming language employs various advanced algorithms and techniques to optimize this process:
-
Sequence alignment algorithms identify suitable target sequences for amplification or detection.
-
Primer/probe design algorithms select appropriate, specific, and efficient oligonucleotide sequences.
-
Secondary structure prediction algorithms prevent non-specific binding or unwanted interactions between oligonucleotides.
-
Simulated annealing or genetic algorithms optimize chemical synthesis and minimize errors or side reactions. C + + programs generate optimized protocols for the design and synthesis of oligonucleotides by integrating these algorithms and techniques.

The interface of the POSoligo program enables users to select a document or input a sequence directly into the "Input" section. Users can adjust the lengths of contiguous long strands (COS) and short patch strands (POS) based on the experiment's objectives and requirements. Clicking on "Calculate" generates the results.
Application
Design of an oligonucleotide set for SARS-CoV-2
During the coronavirus pandemic, we developed this software to combat the virus, aiming to save time and streamline PCR synthesis. To evaluate POSoligo, we designed the nucleotide sequence of the coding region of the S1 protein gene of the SARS-CoV-2 virus (GenBank registry no. QHD43416) encompassing the RBD amino acids Arg319–Lys529, totaling 210 base pairs. Subsequently, we appended the CMV promoter at its 5′-end and the polyA tail at its 3′-end to construct the CMV + RBD + polyA60 expression frame (Fig. 2).

SARS-CoV-2 S protein primary structure and promoter CMV + target antigen RBD + polyA60 complete expression frame. (A) RBD located on the S1 subunit in S protein; (B) promoter CMV + target antigen RBD + polyA60 complete expression frame.
The program generated a total of 34 oligonucleotides (Table 1), including CMV1–CMV10 and RBD1–RBD8 as long-structured oligonucleotides (COS) ranging from 50–120 base pairs, and P1–P7 and CP1–CP9 as short structured POS spanning 22–30 base pairs, respectively. Notably, CMV-R served as an intermediate POS for binding the CMV promoter to the RBD reading frame, while CMV-F and RBD-R were utilized as upstream and downstream primers for PCR amplification post-segment assembly. Additionally, RBD-F and RBD-R functioned as primers for reverse transcription cDNAs, followed by sequencing to validate the correct RBD sequence expression. Furthermore, the software's folder was utilized to predict the secondary structure of COS RNA, ensuring free 5′- and 3′-ends, while minimizing self-annealing in the middle portion, which readily denatures at high temperatures (Fig. 3).

Schematic diagram illustrating the flow of the POS method, with the secondary structure of the POS chain maintaining double ends free in the upper right corner.
Synthesis of SARS-CoV-2 RBD gene in vitro
CMV1-CMV10, RBD1-RBD8, P1-P7, CP1-CP9, and CMV-R (Table 1) were combined using the 2 × Pfu PCR Mix system and subjected to LCR on MG25 + under the following conditions: 95 °C for 5 min, 45 cycles of 95 °C for 30 s, 51 °C for 20 s, and 45 °C for 4 min, with a final overnight incubation at 45 °C for high-temperature ligation.
The PCR reaction mixture comprised 1 μL of LCR product, 1 μL of each CMV-F and RBD-R primer (Table 1), 12.5 μL of 2 × Pfu PCR Mix, and 9.5 μL of ddH2O. PCR amplification followed this program: 94 °C for 3 min, 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 2 min, with a final extension at 72 °C for 5 min. Target bands were identified via 1% gel electrophoresis and excised using a gel recovery kit.
Figure 4 (S1) illustrates the successful in vitro synthesis and amplification of the expression frame sequence of CMV + target antigen RBD + polyA60 (1530 bp) following LCR + PCR reaction. The electrophoresis fragment matched the expected size, confirming the successful synthesis of the expression frame sequence.

1.0% agarose gel electrophoresis to verify 1 μL LCR-PCR product. M: Marker (1 kbp); 1: LCR-PCR product.
Synthesis and validation of RBD gene
The LCR-PCR product was recovered from the gel, and its concentration was measured spectrophotometrically, yielding 0.32 μg/μL. Subsequently, the DNA recovered from the gel was transfected into 293FT cells and incubated for 48 h. The RNA extracted and measured spectrophotometrically at a concentration of 1.3 μg/μL was reverse-transcribed into cDNA. The RT-PCR products were analyzed by electrophoresis, showing a match with the expected size of the RBD target gene fragment (Fig. 5A & S2). Sequencing results confirmed the successful synthesis of the RBD fragment, measuring 639 bp (Fig. 5B & Supplement 3).

(A) 1.0% agarose gel electrophoresis to verify 1 μL cDNA-PCR product. M: Marker (1 kbp); 1, 2: RBD fragments (C1, C2) amplified with reverse transcribed cDNA as the template; 3: negative control (CON). (B) Sequencing results.
Discussion
Our in vitro gene synthesis approach offers numerous advantages compared to many current methodologies and tools. In our previous gene synthesis process, designing a large number of primers using the PCR synthesis method often led to mismatches in repetitive sequences, resulting in shifting or loss of some repetitive sequences in the synthesized whole gene fragment. To reduce the error rate associated with manual primer design for whole gene synthesis, our software generates primers in batch, significantly reducing the mismatch rate and greatly facilitating whole gene synthesis.
Advantage 1: Unlike most modern techniques that require dividing target nucleotides into stable and homogeneous oligonucleotides with consistent thermodynamic properties, such as Gene2Oligo26 and Assembly PCR Oligo Maker28, our software cleaves sequences into varying lengths while specifically maintaining the 5′- and 3′-ends of each POS sequence free and unobstructed. This design greatly facilitates the correct ligation of adjacent structural oligonucleotides by Taq DNA ligase. Unlike techniques that rely on maintaining a consistent Tm value among different oligodeoxynucleotides, our method releases any residual secondary structure during denaturation at 95 °C in the LCR without considering the stability of the Tm value. While Stemmer et al.18 synthesized a 2.7-kbp sequence in a single step, verification during an intermediate stage is necessary due to an increased risk of errors in long sequences, reducing the likelihood of obtaining an accurate fragment after assembly10,11,12. Therefore, shortening long sequences is crucial to improve assembly accuracy. In POSoligo, users can calculate increasingly uniform lengths of oligonucleotides within a defined range using the "segment length" option. Advantage 2: Unlike many current tools that design complete double-stranded DNA using various methods involving special ends, enzymatic methods, or considering different lengths of isolated fragments, our software streamlines the process. It avoids the steep learning curve associated with these tools and instead follows common assembly methods such as PCR and LCR21,22,25,26,29,30,31,32.
POSoligo provides clear output labeling below the sequence alignment once the calculation is completed. Additionally, it generates an "output" file in the root folder, which can be easily viewed using Notepad or similar software. The results can be copied directly into an email or other applications, facilitating communication with the manufacturer of the synthesized sequence. We have also compiled the program in C + + to ensure usability across multiple systems, and we plan to develop a web version of the program in the near future to further enhance accessibility.
POSoligo is highly versatile and can be widely used for designing long DNA fragments in synthetic biology and biotechnology research. We are committed to continuously upgrading the functionality of the software to better serve the needs of researchers.
Data availability
Data is provided within the manuscript or supplementary information files. The software during the current study are available in the [figshare] repository, DOI: https://doi.org/https://doi.org/10.6084/m9.figshare.24879006.v1.
References
Hughes, R. A. & Ellington, A. D. Synthetic DNA synthesis and assembly: Putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 9(1), a023812. https://doi.org/10.1101/cshperspect.a023812 (2017).
Roy, S. & Caruthers, M. Synthesis of DNA/RNA and their analogs via phosphoramidite and H-phosphonate chemistries. Molecules 18(11), 14268–14284. https://doi.org/10.3390/molecules181114268 (2013).
Goeddel, D. V. et al. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc. Natl. Acad. Sci. U. S. A. 76(1), 106–110. https://doi.org/10.1073/pnas.76.1.106 (1979).
Heyneker, H. L. et al. Synthetic lac operator DNA is functional in vivo. Nature 263(5580), 748–752. https://doi.org/10.1038/263748a0 (1976).
Itakura, K. et al. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science 198(4321), 1056–1063. https://doi.org/10.1126/science.412251 (1977).
Tian, J. et al. Accurate multiplex gene synthesis from programmable DNA microchips. Nature 432(7020), 1050–1054. https://doi.org/10.1038/nature03151 (2004).
Fodor, S. P. et al. Light-directed, spatially addressable parallel chemical synthesis. Science 251(4995), 767–773. https://doi.org/10.1126/science.1990438 (1991).
Gao, X. et al. A flexible light-directed DNA chip synthesis gated by deprotection using solution photogenerated acids. Nucleic Acids Res. 29(22), 4744–4750. https://doi.org/10.1093/nar/29.22.4744 (2001).
Patwardhan, R. P. et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol. 30(3), 265–270. https://doi.org/10.1038/nbt.2136 (2012).
LeProust, E. M. et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 38(8), 2522–2540. https://doi.org/10.1093/nar/gkq163 (2010).
Kosuri, S. & Church, G. M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 11(5), 499–507. https://doi.org/10.1038/nmeth.2918 (2014).
Wan, W. et al. Error removal in microchip-synthesized DNA using immobilized MutS. Nucleic Acids Res. 42(12), e102. https://doi.org/10.1093/nar/gku405 (2014).
Saaem, I., Ma, S., Quan, J. & Tian, J. Error correction of microchip synthesized genes using Surveyor nuclease. Nucleic Acids Res. 40(3), e23. https://doi.org/10.1093/nar/gkr887 (2012).
Dormitzer, P. R. et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 5(185), 185ra68. https://doi.org/10.1126/scitranslmed.3006368 (2013).
Klein, J. C. et al. Multiplex pairwise assembly of array-derived DNA oligonucleotides. Nucleic Acids Res. 44(5), e43. https://doi.org/10.1093/nar/gkv1177 (2016).
Au, L. C., Yang, F. Y., Yang, W. J., Lo, S. H. & Kao, C. F. Gene synthesis by a LCR-based approach: High-level production of leptin-L54 using synthetic gene in Escherichia coli. Biochem. Biophys. Res. Commun. 248(1), 200–203. https://doi.org/10.1006/bbrc.1998.8929 (1998).
Dillon, P. J. & Rosen, C. A. A rapid method for the construction of synthetic genes using the polymerase chain reaction. Biotechniques 9(3), 298–300 (1990).
Stemmer, W. P., Crameri, A., Ha, K. D., Brennan, T. M. & Heyneker, H. L. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene 164(1), 49–53. https://doi.org/10.1016/0378-1119(95)00511-4 (1995).
Pusch, C. M., Giddings, I. & Scholz, M. Repair of degraded duplex DNA from prehistoric samples using Escherichia coli DNA polymerase I and T4 DNA ligase. Nucleic Acids Res. 26(3), 857–859. https://doi.org/10.1093/nar/26.3.857 (1998).
Gibson, D. G. et al. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319(5867), 1215–1220. https://doi.org/10.1126/science.1151721 (2008).
Engler, C. & Marillonnet, S. Golden Gate cloning. Methods Mol. Biol. 1116, 119–131. https://doi.org/10.1007/978-1-62703-764-8_9 (2014).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6(5), 343–345. https://doi.org/10.1038/nmeth.1318 (2009).
Gibson, D. G. et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329(5987), 52–56. https://doi.org/10.1126/science.1190719 (2010).
Hoover, D. M. & Lubkowski, J. DNAWorks: An automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res. 30(10), e43. https://doi.org/10.1093/nar/30.10.e43 (2002).
Engler, C. & Marillonnet, S. Combinatorial DNA assembly using Golden Gate cloning. Methods Mol. Biol. 1073, 141–156. https://doi.org/10.1007/978-1-62703-625-2_12 (2013).
Rouillard, J. M. et al. Gene2Oligo: Oligonucleotide design for in vitro gene synthesis. Nucleic Acids Res. 32, W176–W180. https://doi.org/10.1093/nar/gkh401 (2004).
Yang, G. et al. Patch oligodeoxynucleotide synthesis (POS): A novel method for synthesis of long DNA sequences and full-length genes. Biotechnol. Lett. 34(4), 721–728. https://doi.org/10.1007/s10529-011-0832-0 (2012).
Rydzanicz, R., Zhao, X. S. & Johnson, P. E. Assembly PCR oligo maker: A tool for designing oligodeoxynucleotides for constructing long DNA molecules for RNA production. Nucleic Acids Res. 33, W521–W525. https://doi.org/10.1093/nar/gki380 (2005).
Yamazaki, K. I., de Mora, K. & Saitoh, K. BioBrick-based “Quick Gene Assembly” in vitro. Synth. Biol. (Oxf). 2(1), ysx003. https://doi.org/10.1093/synbio/ysx003 (2017).
Smith, H. O., Hutchison, C. A. 3rd., Pfannkoch, C. & Venter, J. C. Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 100(26), 15440–15445. https://doi.org/10.1073/pnas.2237126100 (2003).
Li, M. et al. In vivo production of RNA nanostructures via programmed folding of single-stranded RNAs. Nat. Commun. 9(1), 2196. https://doi.org/10.1038/s41467-018-04652-4 (2018).
Annaluru, N. et al. Assembling DNA fragments by USER fusion. Methods Mol. Biol. 852, 77–95. https://doi.org/10.1007/978-1-61779-564-0_7 (2012).
Funding
The Special fund for cancer prevention and treatment of Shanghai Science and Technology Development Foundation (Grant number CT20200517A).
Author information
Authors and Affiliations
Contributions
Y.Y.T. designed the experiment, participated in the writing of the manuscript and created the images. J.S. completed the experiment and finished the writing of the manuscript, Y.C. participated in the experiment, C.H.Y. supervised and revised the manuscript, H.W. participated in the experiment, C.X.L supervised and revised the manuscript, N.N.D. designed and supervised the study, and G.H.Y. designed and supervised the study, interpreted data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tong, Y., Sun, J., Chen, Y. et al. POSoligo software for in vitro gene synthesis. Sci Rep 14, 11117 (2024). https://doi.org/10.1038/s41598-024-59497-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59497-3
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
