
Abstract
Bimetallic FePt nanoparticles with L1 0 structure are attracting a lot of attention due to their high magnetocrystalline anisotropy and high coercivity what makes them potential material for storage of ultra-high density magnetic data. FePt nanoclusters are considered also as nanocatalysts for growth of carbon nanotubes of different chiralities. Using the DFT-LCAO CRYSTAL14 code, we have performed large-scale spin-polarized calculations on 19 different polyhedral structures of FePt nanoparticles in order to estimate which icosahedral or hcp-structured morphology is the energetically more preferable. Surface energy calculations of all aforementioned nanoparticles indicate that the global minimum corresponds to the nanocluster possessing the icosahedron “onion-like” structure and Fe43Pt104 morphology where the outer layer consists of Pt atoms. The presence of the Pt-enriched layer around FePt core explains high oxidation resistance and environmental stability, both observed experimentally.
Similar content being viewed by others

Introduction
Magnetic nanoparticles (NPs) with sizes ranging from 2 to 20 nm represent an important class of artificial nanostructured materials. Their magnetic properties essentially depend on the NP size because the thermal energy kT becomes comparable to the KV product term, where k T, K and V are the Boltzmann constant, the temperature, the constant of the so-called magnetic anisotropy of the nanoparticle (NP) and its volume, respectively1. As a result, the magnetization of the nanocluster can randomly flip direction depending on the temperature, and thus NP can be fixed in the so-called superparamagnetic state1,2,3.
FePt bulk possesses L1 0 4 chemically ordered P4/mmm tetragonal structure or chemically disordered A1 (\(Fm\overline{3}m\)) cubic structure2, 5. It was shown recently that the atomic ratio of Fe and Pt in Fe x Pt1–x nanoparticles (NPs) synthesized by the sol-gel method (where x was changed between 0.3 and 0.8) plays an essential role for the structural and magnetic properties of these NPs6. FePt NPs possessing a near-stoichiometric atomic percentage of Fe and Pt belong to the important class of magnetic nanomaterials. FePt L1 0 NPs have attracted considerable attention because of their extremely high magnetic anisotropy7 making them especially useful for practical applications in solid-state devices, e.g., in high-density magnetic recording media8, 9, and in biomedicine, e.g., as contrast agents for the magnetic resonance imaging10 or as the basis for neutron activated coating when annealing the FePt core–shell NPs for cancer treatment11.
Magnetic properties of NPs also allow one to use them as a catalyst for the growth of carbon nanotubes (CNTs) with predictable chiralities12. Chirality of an arbitrary CNT depends on the direction of an external magnetic field while diameter depends on the size of FePt NP2. It should be also noted that FePt nanoparticles are chemically more stable than high-magnetic nanoclusters of Co and Fe, as well as other high coercive materials like CoSm5 and Nd2Fe14B2. Synthesized L1 0 FePt/MnFe2O4 core-shell nanocomposites also possess attracting magnetic properties13.
FePt NPs were mainly synthesized using organometallic chemistry7, 14 or gas-phase condensation and nucleation methods15,16,17. The latter can be applied for the synthesis of NPs with various shapes: cuboctahedrons, icosahedrons, and faceted spheres17, 18.
A number of different FePt NP sectioning morphologies and properties simulated using first-principles calculations are described in the literature19,20,21,22,23,24,25,26,27,28. Free energies of (100), (001), (110), (011) and (111) flat sections of L1 0 FePt bulk were calculated to estimate surface energy anisotropy of FePt NPs19. Evidently, {111} facets of nanocrystal were found to be energetically the most favorable. This result is important for analysis of high-resolution transmission electron microscopy images, since FePt NPs do not show obvious surface faceting on the early stage of annealing. It means that some structural features are rather depending on the kinetic factors than on the free surface energies. In ref. 20, Gruner has shown that for L1 0 ordered alloys, the two (001) surfaces perpendicular to the shortened c-axis are preferably covered by Pt atoms analogously to the outermost shell of Fe144Pt165 NP completely covered by Pt.
The systematic survey of the size dependence of the energetic order and magnetic properties of FePt nanoparticles of various morphologies was performed by Gruner et.al.21, 22. Low-indexed surfaces of mono-metallic Fe, Co, and Pt, as well as bimetallic, ordered FePt, CoPt, and MnPt NPs were calculated using the density-functional theory (DFT)23. These calculations showed that elemental, Pt-covered surfaces are preferable over Fe and Co covered and mixed surfaces of the same orientation. The first-principles DFT calculations were used to determine how Pt surface segregation (exchanging interior Pt with surface Fe atoms) would affect the magnetic properties of L1 0 ordered FePt NPs28. In ref. 29, the structural stability and structural features of FePt NPs were studied using Monte Carlo algorithms generating models of different shapes, Fe/Pt ratios and atomic compositions. It was shown that the icosahedron possesses the best structural stability and the lowest energy in comparison with all other shapes of NPs (truncated octahedron, octahedron, decahedron, or hexahedron).
Despite these studies it is not precisely known at this moment what types of particles are the most stable and which types can be obtained experimentally. The growth mechanism of NPs is still being discussed, but the recent studies bring evidences that they exhibit shell periodicity and grow by accretion of atomic layers. Still, there are many known NPs that possess shell-core structure. For example, such a type of bimetallic NPs have been simulated recently30, e.g., Pd m Au n 31. The method of topological energy expression (TOP) applied for them has enabled one to determine the most energetically stable atomic arrangements by global optimization of the mutual positions of different atoms (or chemical ordering). As in the case of FePt core-shell NPs, the energetically more preferable configurations were found to be fully covered by noble metal atoms (Au).
In order to shed more light on the NP surface structure and the mechanism of NP growth, we have performed a series of DFT calculations using the CRYSTAL14 code32. Using the methodology suggested in ref. 33 we have calculated the Gibbs free energy to determine the lowest-energy structure of a NP surface as well as the morphology and stoichiometry of FePt nanocluster which correspond to the thermodynamically favorable surface.
The paper is organized as follows. In the first subsection of “Methods” section the computational details of ab initio calculations by a linear combination of atomic orbitals (LCAO) within the DFT approach used in our modelling are given. The second subsection of “Methods” section describes different atomistic models of icosahedral and hexagonal close-packed (hcp) FePt NPs used in calculations. The obtained results are analyzed in the “Results” section divided into three Subsections. The “Bulk calculations” subsection contains detailed information about Fe, Pt and FePt bulk crystal structure calculations. In the “Magnetic properties of FePt nanoparticles” subsection, we analyze the magnetic structure of the considered NPs. The “Thermodynamical analysis of FePt nanoparticles structures” subsection presents the thermodynamical analysis of FePt NPs. Finally, the conclusions are given in the “Summary” section.
Methods
Computational details
Spin-polarized large-scale LCAO calculations on FePt NPs have been performed using the CRYSTAL14 code32 within the DFT-PWGGA exchange-correlation functional34,35,36. For the Fe, the all-electron atomic centered Gaussian-type function basis set (BS) has been adopted in the triple-zeta valence form37, while for the Pt, we have used the effective core pseudopotential BS of 311s-1sp-221p-41d 38, with the exponents of core and valence shells being unchanged. Unique options provided by the CRYSTAL code allowed us to perform a consistent comparison between the HF and DFT descriptions of the atomic and electronic properties of molecules and solids, including performance of the hybrid exchange-correlation functionals. With respect to the plane-waves codes the CRYSTAL code allows us to avoid artificial periodicity of repeated 3D boxes and perform simulations for stand-alone nanoparticle in the most efficient manner.
The threshold parameters of CRYSTAL code (ITOLn) for evaluation of different types of bielectronic integrals (overlap and penetration tolerances for Coulomb integrals, ITOL1 and ITOL2, overlap tolerance for exchange integrals ITOL3, as well as pseudo-overlap tolerances for exchange integral series, ITOL4 and ITOL532) have been set to 6, 6, 6, 6, and 12, respectively. If the overlap between the two atomic orbitals is smaller than 10−ITOLn, the corresponding integral is truncated. Further increase of threshold parameters results in much more expensive calculations yielding only a negligible gain in the total energy (10−7 a.u.). Calculations are considered as converged when the total energy obtained in the self-consistent field procedure differs by less than 10−7 a.u. in two successive cycles. Full geometry optimization has been performed for each considered model.
Models of nanoclusters
A number of FePt nanoclusters with different stoichiometry and atom arrangement has been set up with preserved L1 0 structure. There have been icosahedra and hcp configurations, possessing either layered or “onion-like” structures. The latter are observed and described, e.g., in ref. 39.
We have observed that the icosahedral structures formed the so-called “magic number” clusters22, where the number of atoms N is given as a function of the number n of closed geometric shells
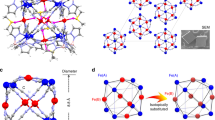
In this study, all chosen icosahedra were of the same size −147 atoms40, which corresponds to a diameter of ≈1.6 nm and number of closed atomistic shells n = 3 (Fig. 1). Analogous hcp structures consist of 153 atoms40, which correspond to a diameter of ≈1.4 nm (Fig. 2). In this study, we have chosen NPs of icosahedron and hcp structure as both contain the largest number of the most stable {111} facets. At the same time both icosahedron17 and hcp 41 structured FePt NPs were observed experimentally.

Selected icosahedral cluster models with initial morphology.

Selected hcp nanocluster models with initial morphology.
For both types of NPs possessing icosahedral and hcp configurations, we have constructed models with “onion-like” atom arrangement, which are isotropic in contrast with layered and other types of arrangements. In total, we have considered and calculated 19 NP structures. Two “onion-like” (Fe43Pt104 and Fe104Pt43), and three layered icosahedron structures (Fe76Pt71, Fe71Pt76, Fe79Pt68), each containing 147 atoms, are presented in Fig. 1.
Two “onion-like” hcp structures (Fe45Pt108 and Fe108Pt45), and twenty layered hcp structures (Fe82Pt71, Fe71Pt82, Fe81Pt72, Fe72Pt81, Fe80Pt73, Fe73Pt80, Fe79Pt74, Fe74Pt79, Fe78Pt75, Fe75Pt78, Fe77Pt76, Fe76Pt77), containing 153 atoms, are collected in Fig. 2.
Results
Bulk calculations
Properties of Fe, Pt and FePt 3D crystals have been calculated at the beginning in order to obtain energy reference for calculations of chemical potentials of these compounds as well as to check the quality of chosen BSs. For this purpose, we have calculated such Fe, Pt and FePt bulk properties as lattice constants, magnetic moments of atoms, and bulk moduli comparing these values with the experimental data reported in the literature.
The bulk phase of L1 0 FePt has been calculated for P4/mmm space group that gives lattice constants of 2.743 Å and 3.780 Å as well as magnetic moments equal to 3.144 μ B and 0.169 μ B for Fe and Pt, respectively. Fe bulk has been calculated for \(Im\overline{3}m\) space group yielding a 0 = 2.812 Å and 2.140 μ B. For Pt bulk calculations, the \(Fm\overline{3}m\) space group has been adopted that yields lattice constant of 4.029 Å. (The experimental lattice constants are equal to 2.86 Å for Fe42, 3.92 Å for Pt43, and a = 2.7301 Å and c = 3.7879 Å for FePt ordered structure44).
We have also calculated bulk moduli for Fe, Pt and FePt bulk, obtaining values B0Fe = 220 GPa, B0pt = 218 GPa and B0FePt = 202 GPa, correspondingly. These results qualitatively agree with the existing experimental data: Fe bulk modulus measured at 300 K is equal to B0Fe = 166.2 GPa45 while Pt experimental bulk modulus at 300 K is equal to B0Pt = 280 GPa46. Finally, a bulk modulus for FePt polycrystalline L1 0 structure recently measured at room temperature is equal to B0FePt = 208.1 GPa47.
Magnetic properties of FePt bulk and nanoparticles
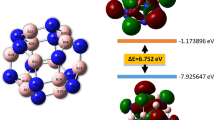
The distribution of magnetic moments of Fe and Pt atoms has been analyzed for all considered NP structures. Magnetic moments M Fe and M Pt of Fe and Pt atoms inside the studied FePt NPs, which depend on the the distance between the corresponding atom and the NP center, are shown in Fig. 3 for four selected models of FePt NPs. The mean magnetic moments of Fe and Pt atoms for seven of the studied FePt NP models as well as for the FePt bulk are shown in Fig. 4. It has been found that the mean M Fe in each NP is similar to M Fe in bulk phase, while the mean M Pt in NPs is about 20% higher. In all cases magnetic moments of Fe atoms grow with the distance away from the center of NP. The trend of the magnetic moment distribution for other NP models which are not shown in Figs 3–4 is similar. These values qualitatively agree with the existing experimental data for FePt NPs: ≈2.2–2.6 μ B 48,49,50 for Fe atoms and ≈ 0.4 μ B for Pt atoms48. Figure 3 also shows that Fe43Pt104 nanoparticle possesses high symmetry (C2h ), so large numbers of Fe and Pt atoms are located at equal distance from the center.

Magnetic moments of Fe and Pt atoms for some studied FePt NPs (icosahedral “onion-like” Fe43Pt104 NP, icosahedral layered Fe79Pt68 NP, hcp “onion-like” Fe45Pt108 NP and hcp layered Fe71Pt82 NP) depending on the distance between the constructed NP center and the selected atom. Values are given in μ B.

Mean magnetic moments of Fe and Pt atoms for some studied FePt NPs in μ B (from left to right): icosahedron “onion-like” Fe43Pt104 and Fe104Pt43 and layered Fe79Pt68 structures, hcp “onion-like” Fe45Pt108 and Fe108Pt45, and layered Fe71Pt82 and Fe81Pt72 structures, as well as magnetic moments of Fe and Pt atoms in FePt bulk calculations.
Thermodynamical analysis of FePt NP structures
In order to predict the most likely particle shape and composition, the detailed knowledge of the surface free energies G of the competing NP surface’s morphologies and internal interfaces is needed. The thermodynamic approach used in the current study in order to estimate the stability of FePt NP surfaces has been adopted from the refs 23 and 33. According to the prescription given in the ref. 33, we assume that the stable NP surface has to be in equilibrium with the FePt bulk phase. Therefore, the most stable NP has the lowest surface Gibbs free energy defined as
where t indicates the NP morphology (stoichiometry), A the NP surface area, N i the number of atoms of type i in the NP \({E}_{t}^{{\rm{FePt}}}\) is the total energy of a NP with t morphology and \({E}_{{\rm{bulk}}}^{{\rm{FePt}}}\) is the FePt total energy in the L1 0 bulk phase, while \({\rm{\Delta }}{\mu }_{i}={\mu }_{i}-{E}_{{\rm{bulk}}}^{i}\), (i = Fe, Pt) are deviations of chemical potentials for metal atoms from their energy values in the bulk phase. Since pV term (V is unit cell volume) and the differences in vibrational Gibbs free energy between the bulk solid and a corresponding NP is negligibly small33, we omit these two contributions. This allows us to replace the Gibbs free energies in eq. (2) by the internal energies U calculated from the first principles.
In order to avoid the precipitation of Fe or Pt at NP surface, as well as to prevent metal atoms from leaving the NP, the following conditions must be satisfied:
where \({E}_{{\rm{FePt}}}^{f}\) is the calculated formation energy of −0.48 eV for FePt bulk vs. −0.24 eV in the experiment51. Calculated formation energies for Fe and Pt bulk are −6.49 and −5.60 eV vs. −4.28 and −5.84 eV obtained from the experiment, respectively51. Negative formation energies were calculated with respect to the ground state energies of free standing atoms.
Thermodynamic stability diagram is constructed in Fig. 5 based on eqs (2 and 3). We can predict from it that the surface of Pt-covered Fe43Pt104 “onion-like” NP is the most stable, in agreement with a recent experimental observation17. External shells of experimentally studied FePt nanoparticles were found to be Pt-rich as energetically more stable because surface may suppress undesired magnetic exchange coupling. For more convincing conclusion on morphology of FePt NPs, we have to consider nanoparticles with diameter at least 5–6 nm, as synthesized in the aforementioned study17 that is beyond our current computational facilities. Nevertheless, we expect that our model of Pt-covered nanoparticle with additional 2 or 4 shells (561 and 1415 atoms, respectively) and vacancy formation at the edges of the outer shell would allow additional decrease of its free surface energy, thus leading to realistic description of magnetic FePt nanoparticles.

Thermodynamic stability diagram as a function of Pt chemical potential built for all FePt NPs under study. Diagram takes into account precipitation conditions for both Fe and Pt metals.
Summary
In this study, we have performed large-scale DFT calculations of FePt nanoparticles of different shapes. Our calculations show that the average magnetic moment of Fe and Pt atoms does not change significantly when comparing it for bulk FePt structure and Fe43Pt104 cluster (e.g., M Fe,av = 3.17 μ B and M Pt,av = 0.21 μ B for Fe43Pt104 particle vs. M Fe,av = 3.14 μ B and M Pt,av = 0.17 μ B for FePt bulk phase). Using thermodynamical approach, we have found that the global minimum of surface energy corresponds to nanocluster with icosahedron “onion-like” structure and Fe43Pt104 morphology where the outer layer consists of Pt atoms only, which is in a good agreement with results obtained elsewhere17. This nanoparticle can be used for further simulations of enlarged cluster and adsorption of regular network of C atoms upon it resulting in a growth of carbon nanotubes.
References
Xu, C. & Sun, S. Monodisperse magnetic nanoparticles for biomedical applications. Polym. Int. 56, 821–826, doi:10.1002/pi.2251 (2007).
Sun, S. Recent advances in chemical synthesis, self-assembly, and applications of FePt nanoparticles. Adv. Mat. 18, 393–403, doi:10.1002/adma.200501464 (2006).
Adelani, P. O., Duke, A. N., Zhou, B. H. & Rinehart, J. D. Role of magnetic concentration in modulating the magnetic properties of ultra-small FePt nanoparticles. Inorganica Chimica Acta 460, 114–118, doi:10.1016/j.ica.2016.09.020 (2017).
Laughlin, D. E., Srinivasan, K., Tanase, M. & Wang, L. Crystallographic aspects of L10 magnetic materials. Scripta Materialia 53, 383–388, doi:10.1016/j.scriptamat.2005.04.039 (2005).
Gutfleisch, O., Lyubina, J., Müller, K.-H. & Schultz, L. FePt hard magnets. Adv. Eng. Mat. 7, 208–212, doi:10.1002/adem.200400183 (2005).
Liu, Y. et al. Structural and magnetic properties of the ordered FePt3, FePt and Fe3Pt nanoparticles. J. Solid State Chem. 209, 69–73, doi:10.1016/j.jssc.2013.10.027 (2014).
Sun, S., Murray, C. B., Weller, D., Folks, L. & Moser, A. Monodisperse FePt nanoparticles and ferromagnetic FePt nanocrystal superlattices. Sci. 287, 1989–1992, doi:10.1126/science.287.5460.1989 (2000).
Christodoulides, J. A. et al. CoPt and FePt thin films for high density recording media. J. Appl. Phys. 87, 6938–6940, doi:10.1063/1.372892 (2000).
Weller, D. et al. High K u materials approach to 100 Gbits/in2. Magn. IEEE Transactions 36, 10–15, doi:10.1109/20.824418 (2000).
Maenosono, S., Suzuki, T. & Saita, S. Superparamagnetic FePt nanoparticles as excellent MRI contrast agents. J. Magn. Magn. Mat. 320, L79–L83, doi:10.1016/j.jmmm.2008.01.026 (2008).
Seemann, K. et al. Magnetic heating properties and neutron activation of tungsten-oxide coated biocompatible FePt core-shell nanoparticles. J. Control. Release 197, 131–137, doi:10.1016/j.jconrel.2014.11.007 (2015).
Bellucci, S. et al. Memory nanodevices based on carbon nanotube-FePt interconnects: Electromagnetic simulations and magnetically stimulated nanotube growth. J. Mat. Sci. Eng. B 5, 120–134, doi:10.17265/2161-6221/2015.3-4.003 (2015).
Akbari, H., Zeynali, H., Sebt, S. A. & Anjami, A. Magnetic and structural properties of L10 FePt/MnFe2O4 nanocomposites. Superlattices and Microstructures 69, 99–107, doi:10.1016/j.spmi.2014.02.004 (2014).
Elkins, K. E. et al. Ultrafine FePt nanoparticles prepared by the chemical reduction method. Nano Lett. 3, 1647–1649, doi:10.1021/nl034734w (2003).
Stappert, S., Rellinghaus, B., Acet, M. & Wassermann, E. F. Gas-phase preparation of L10 ordered FePt nanoparticles. J. Cryst. Growth 252, 440–450, doi:10.1016/S0022-0248(03)00935-7 (2003).
Qiu, J.-M. & Wang, J.-P. Monodispersed and highly ordered L10 FePt nanoparticles prepared in the gas phase. Appl. Phys. Lett. 88, 192505, doi:10.1063/1.2202130 (2006).
Wang, R. et al. Layer resolved structural relaxation at the surface of magnetic FePt icosahedral nanoparticles. Phys. Rev. Lett. 100, 017205, doi:10.1103/PhysRevLett.100.017205 (2008).
Wang, R. et al. FePt icosahedra with magnetic cores and catalytic shells. J. Phys. Chem. C 113, 4395–4400, doi:10.1021/jp811280k (2009).
Hong, S. & Yoo, M. H. Surface energy anisotropy of FePt nanoparticles. J. Appl. Phys. 97, 084315, doi:10.1063/1.1863424 (2005).
Gruner, M. E. Antiferromagnetism and segregation in cuboctahedral FePt nanoparticles. J. Phys. D: Appl. Phys. 41, 134015, doi:10.1088/0022-3727/41/13/134015 (2008).
Gruner, M., Rollmann, G., Entel, P. & Farle, M. Multiply twinned morphologies of FePt and CoPt nanoparticles. Phys. Rev. Lett. 100, 087203, doi:10.1103/PhysRevLett.100.087203 (2008).
Entel, P. & Gruner, M. E. Large-scale ab initio simulations of binary transition metal clusters for storage media materials. J. Phys.: Condens. Matter 21, 064228, doi:10.1088/0953-8984/21/6/064228 (2009).
Dannenberg, A., Gruner, M., Hucht, A. & Entel, P. Surface energies of stoichiometric FePt and CoPt alloys and their implications for nanoparticle morphologies. Phys. Rev. B 80, 245438, doi:10.1103/PhysRevB.80.245438 (2009).
Gruner, M. E. Chemical trends in structure and magnetism of bimetallic nanoparticles from atomistic calculations. J. Phys. D: Appl. Phys. 43, 474008, doi:10.1088/0022-3727/43/47/474008 (2010).
Gruner, M. E. & Entel, P. Competition between ordering, twinning, and segregation in binary magnetic 3d-5d nanoparticles: A supercomputing perspective. Int. J. Quantum Chem. 112, 277–288, doi:10.1002/qua.23254 (2012).
Li, Y., Qi, W., Huang, B. & Wang, M. Modeling the size- and shape-dependent surface order-disorder transition of Fe0.5Pt0.5 nanoparticles. J. Phys. Chem. C 116, 26013–26018, doi:10.1021/jp3065079 (2012).
Cuadrado, R. & Chantrell, R. Electronic and magnetic properties of bimetallic L10 cuboctahedral clusters by means of fully relativistic density-functional-based calculations. Phys. Rev. B 86, 224415, doi:10.1103/PhysRevB.86.224415 (2012).
Lv, H., Lei, Y., Datta, A. & Wang, G. Influence of surface segregation on magnetic properties of FePt nanoparticles. Appl. Phys. Lett. 103, 132405, doi:10.1063/1.4822172 (2013).
Fan, T.-E., Liu, T.-D., Zheng, J.-W., Shao, G.-F. & Wen, Y.-H. Structure and stability of Fe-Pt bimetallic nanoparticles: Initial structure, composition and shape effects. J. Alloy. Compd. 685, 1008–1015, doi:10.1016/j.jallcom.2016.06.281 (2016).
Kozlov, S. M., Kovacs, G., Ferrando, R. & Neyman, K. M. How to determine accurate chemical ordering in several nanometer large bimetallic crystallites from electronic structure calculations. Chem. Sci. 6, 3868–3880, doi:10.1039/C4SC03321C (2015).
Kovacs, G., Kozlov, S. M. & Neyman, K. M. Versatile optimization of chemical ordering in bimetallic nanoparticles. J. Phys. Chem. C 121, 10803–10808, doi:10.1021/acs.jpcc.6b11923 (2017).
Dovesi, R. et al. Crystal14: A program for the ab initio investigation of crystalline solids. Int. J. Quantum Chem. 114, 1287–1317, doi:10.1002/qua.24658 (2014).
Reuter, K. & Scheffler, M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure. Phys. Rev. B 65, 035406, doi:10.1103/PhysRevB.65.035406 (2001).
Perdew, J. P. & Wang, Y. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 33, 8800–8802, doi:10.1103/PhysRevB.33.8800 (1986).
Perdew, J. P. & Yue, W. Erratum: Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 40, 3399, doi:10.1103/PhysRevB.40.3399 (1989).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249, doi:10.1103/PhysRevB.45.13244 (1992).
Peintinger, M. F., Oliveira, D. V. & Bredow, T. Consistent gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 34, 451–459, doi:10.1002/jcc.23153 (2013).
Doll, K. CO adsorption on the Pt(111) surface: a comparison of a gradient corrected functional and a hybrid functional. Surf. Sci. 573, 464–473, doi:10.1016/j.susc.2004.10.015 (2004).
Feldman, Y., Zak, A., Popovitz-Biro, R. & Tenne, R. New reactor for production of tungsten disulfide hollow onion-like (inorganic fullerene-like) nanoparticles. Solid State Sci. 2, 663–672, doi:10.1016/S1293-2558(00)01070-0 (2000).
Weigend, F. & Ahlrichs, R. Quantum chemical treatments of metal clusters. Philos. Trans. Royal Soc. London A: Math. Phys. Eng. Sci. 368, 1245–1263, doi:10.1098/rsta.2009.0268 (2010).
Berlanga-Ramarez, E. et al. Magnetism in segregated bimetallic CoRh nanoclusters. Phys. B: Condens. Matt. 354, 278–281, doi:10.1016/j.physb.2004.09.063 (2004).
Pietrokowsky, P. Lattice parameter of alpha iron by divergent beam diffraction. J. Appl. Phys. 37, 4560–4571, doi:10.1063/1.1708081 (1966).
Arblaster, J. W. Crystallographic properties of platinum. Platinum Met. Rev. 41, 12–21 (1997).
Ravindran, P. et al. Large magnetocrystalline anisotropy in bilayer transition metal phases from first-principles full-potential calculations. Phys. Rev. B 63, 144409, doi:10.1103/PhysRevB.63.144409 (2001).
Adams, J. J., Agosta, D. S., Leisure, R. G. & Ledbetter, H. Elastic constants of monocrystal iron from 3 to 500 k. J. Appl. Phys. 100, 113530, doi:10.1063/1.2365714 (2006).
Macfarlane, R., Rayne, J. & Jones, C. Anomalous temperature dependence of shear modulus c44 for platinum. Phys. Lett. 18, 91–92, doi:10.1016/0031-9163(65)90659-1 (1965).
Nakamura, N., Yoshimura, N., Ogi, H. & Hirao, M. Elastic constants of polycrystalline L10-FePt at high temperatures. J. Appl. Phys. 114, 093506, doi:10.1063/1.4819974 (2013).
Antoniak, C. et al. Enhanced orbital magnetism in Fe50Pt50 nanoparticles. Phys. Rev. Lett. 97, 117201, doi:10.1103/PhysRevLett.97.117201 (2006).
Dmitrieva, O. et al. Magnetic moment of Fe in oxide-free FePt nanoparticles. Phys. Rev. B 76, 064414, doi:10.1103/PhysRevB.76.064414 (2007).
Antoniak, C. et al. Inhomogeneous alloying in FePt nanoparticles as a reason for reduced magnetic moments. J. Phys.: Condens. Matt. 21, 336002, doi:10.1088/0953-8984/21/33/336002 (2009).
Chase, M. W. NIST-JANAF thermochemical tables (Am. Chem. Soc., Washington, DC, 1998).
Acknowledgements
Financial support provided by Scientific Research Project for Students and Young Researchers Nr. SJZ/2016/17 implemented at the Institute of Solid State Physics, University of Latvia, is greatly acknowledged. A.P. and R.E. express their gratitude to High-performance computer centers of ISSP (University of Latvia) and St. Petersburg University. This research was partially supported by Graphene Flagship GrapheneCore1-AMD-696656-4.
Author information
Authors and Affiliations
Contributions
D.B., Y.Z., R.E. and S.B. designed and organized the research; A.P. and S.P. constructed models (Figures 1,2) and performed calculations; A.P., S.P. and D.B. constructed the plots (Figures 3–5) and wrote the major part of the text; all authors contributed to the interpretation of the results and editing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Platonenko, A., Piskunov, S., Bocharov, D. et al. First-principles calculations on Fe-Pt nanoclusters of various morphologies. Sci Rep 7, 10579 (2017). https://doi.org/10.1038/s41598-017-11236-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11236-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
