
Abstract
Acute and chronic coronary syndromes (ACS and CCS) are leading causes of mortality. Inflammation is considered a key pathogenic driver of these diseases, but the underlying immune states and their clinical implications remain poorly understood. Multiomic factor analysis (MOFA) allows unsupervised data exploration across multiple data types, identifying major axes of variation and associating these with underlying molecular processes. We hypothesized that applying MOFA to multiomic data obtained from blood might uncover hidden sources of variance and provide pathophysiological insights linked to clinical needs. Here we compile a longitudinal multiomic dataset of the systemic immune landscape in both ACS and CCS (n = 62 patients in total, n = 15 women and n = 47 men) and validate this in an external cohort (n = 55 patients in total, n = 11 women and n = 44 men). MOFA reveals multicellular immune signatures characterized by distinct monocyte, natural killer and T cell substates and immune-communication pathways that explain a large proportion of inter-patient variance. We also identify specific factors that reflect disease state or associate with treatment outcome in ACS as measured using left ventricular ejection fraction. Hence, this study provides proof-of-concept evidence for the ability of MOFA to uncover multicellular immune programs in cardiovascular disease, opening new directions for mechanistic, biomarker and therapeutic studies.
Similar content being viewed by others

Main
Myocardial ischemia is a major driver of mortality and morbidity worldwide1. This is caused by atherosclerosis in coronary arteries, which is clinically subdivided into stable chronic coronary syndromes (CCS) and acute coronary syndromes (ACS). Myocardial infarction (MI), the most severe form of ACS, is primarily caused by an acute disruption of blood flow to the myocardium due to plaque rupture in preexisting CCS2. Local and systemic immune responses are a main driver of atherosclerosis and contribute to thrombosis as well as myocardial remodeling after acute myocardial ischemia3,4. However, the immunological signatures in these disease entities in humans remain incompletely understood.
Single-cell omics approaches allow for the characterization of immune signatures in an unbiased way with unprecedented resolution5. In basic research, these have been used to profile immune cells in atherosclerotic plaques6 and at sites of MI7. Single-cell genomics is increasingly applied in clinical settings8,9, and its diagnostic potential has been shown for oncological disorders10 but not probed in coronary syndromes (CS).
Multiomic factor analysis (MOFA) provides an unsupervised approach for data exploration across multiple data types, enabling the identification of major axes of variation composed of multiple molecular features linking these with underlying molecular processes11. In contrast to analyzing the predictive potential of single variables, this data-driven dimensionality reduction allows for the identification of integrative factors, while retaining the wealth of information contained in the multiomic dataset.
We hypothesized that the concept of applying MOFA on patient blood samples might allow us to define multicellular immune signatures in CS and link these signatures to disease state and outcome. Using a prospective multiomics strategy with an independent second-center validation dataset, we characterize circulating immune signatures and their time course in human CS at the patient level and single-cell resolution.
Results
Baseline characterization of immune profiles in CS
We analyzed the human immune response to myocardial ischemia in a cohort of patients who presented at the cardiology department of the Munich University Hospital (Ludwig-Maximilian University (LMU) Munich (Munich cohort: M)). Patients with ACS were included when presenting with acute ST-elevation MI (STEMI) (see Methods for the inclusion and exclusion criteria and Supplementary Table 1 for the cohort description). Patients with ACS in the Munich cohort were sampled longitudinally to capture all major phases of the immune response during MI3,4 at four sampling timepoints (TPM; Methods). The Munich control cohort, without acute coronary ischemia, further allowed comparison between patients with diagnostically secured CCS and patients with CCS ruled out (non-CCS; Supplementary Table 1). In addition, we validated the key immune signatures using a single-cell RNA sequencing (scRNA-seq) dataset in a second independent cohort of patients with ACS presenting at the University Medical Center Groningen (UMCG; Groningen cohort) measured at three sampling timepoints (TPG; Methods). The Groningen cohort has been primarily sampled, analyzed and reported here12. In the Groningen control cohort, none of the study participants had clinically suspected CCS (corresponding to the Munich non-CCS cohort, further described in Supplementary Table 2).
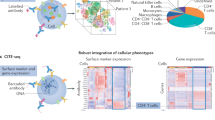
Overall, we enrolled data from a combined total of 117 individuals: comprising samples from n = 62 patients from the Munich cohort (analyzing clinical blood tests, scRNA-seq, cytokine multiplex data, plasma proteomics and neutrophil prime sequencing (prime-seq)) and n = 55 patients from the external validation cohort (Groningen cohort, analyzing scRNA-seq and clinical data). A total of n = 838 individual modality samples (all patient–timepoint combinations from independent omic modalities) were analyzed separately by multiomic or clinical measures (Fig. 1a,b and Methods). From hereon, we refer to a ‘sample’ as the multiomic profile of an individual patient at one specific timepoint.

a, Study design. In the Munich cohort, blood was analyzed from patients with ACS (total of n = 28; total of four timepoints, TP1M–TP4M), patients with CCS (n = 16) and patients with no CCS (n = 18; single timepoint, TP0M). A joint multiomic dataset was created from the Munich cohort by including clinical blood tests (cl), scRNA-seq (SC), flow cytometry, cytokine assay (cy), and plasma proteomics (p) and neutrophil (pmn) prime-seq. This was followed by data integration, MOFA model estimation (Y, input data matrices from each data modality; W, resulting weight matrix; Z, resulting matrix of factor values for each sample) and subsequent downstream analysis such as factor analysis, pathway enrichment, cell–cell communication and prediction. Findings from the Munich data cohort were evaluated in the Groningen data (V2 chemistry) as an independent validation cohort in which blood was analyzed longitudinally from patients with ACS (total of three timepoints TP1G–TP3G, total of n = 24 patients) as well as from a control group (TP0G, n = 31). Created with BioRender.com. b, X-ray images of a coronary catheterization of a patient with ACS: occlusion of the left circumflex artery, indicated by red arrow (left image); intervention, stent implantation (middle image); reperfusion (right image). c, Clinical blood tests. Individual timepoints for sterile ACS (TP1–TP4) compared with those for CCS (TP0M). Mean ± s.e.m. values are shown. d, UMAP plot of scRNA-seq data from PBMCs showing cell-type clusters used for subsequent analyses (n = 148,275). e, Analysis of CLR-transformed cell type abundance based on the scRNA-seq dataset. Data are shown using box–whisker plots (box, median and 25th to 75th percentile; whiskers, minimum to maximum). f, MOFA. Variance decomposition showing the percentage of explained variance per view and factor of the MOFA model with 20 factors. For each view, the heatmap shows the percentage of the variance that is explained by the respective factor. The color coding on the left indicates the data type of each view: green, plasma proteomics; blue, neutrophil prime-seq; orange, cytokine measurements; dark orange, clinical values; purple, scRNA-seq data. The greyscale grading in the heatmap depicts the percentage of variance. The bar plot (right) shows the total percentage of explained variance by all 20 factors. In c and e, parametric-distributed data were analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; nonparametric-distributed data were analyzed using the Kruskal–Wallis test with correction for multiple comparisons by Dunn’s test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. In cases in which only the ordinary one-way ANOVA or Kruskal–Wallis test, but not the multiple comparison, was significant, graphs are marked with a vertical bar on top. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
First, we focused on Munich patients with ACS and a classical acute symptom onset, instant recanalization and no infectious complications during their disease course within the hospital (sterile ACS) and compared them with patients with CCS (see ‘Munich cohort: ethics and patient cohort’ in Methods). The analysis of laboratory values (creatine kinase (CK), creatine kinase MB (CK-MB) isoenzyme and troponin T) confirmed the classical course of acute myocardial ischemia. Dynamic C-reactive protein (CRP) and leukocyte counts confirmed a systemic immune response (Fig. 1c and Extended Data Fig. 1a). Flow-cytometry-based phenotyping revealed no major changes in centered log ratio (CLR) transformed cell type abundances of large classical circulating leukocyte populations in ACS compared with CCS—solely a gradual drop in less frequent immune cells such as dendritic cells, nonclassical monocytes and natural killer (NK) cells (Extended Data Fig. 1b, Supplementary Fig. 1a and Supplementary Table 3). However, although it allows a particularly high resolution of rarer subsets, flow cytometry only identifies predefined classical immune cell populations. To allow for an unsupervised detection of leukocyte subsets, we made use of scRNA-seq (Fig. 1d, Supplementary Fig. 2a,b and Supplementary Tables 4 and 5). Compared with flow cytometry, the granularity of scRNA-seq-defined unsupervised immune cell subsets revealed compositional shifts in immune subsets of T cells and classical monocytes across the disease course of MI (Fig. 1d,e, Extended Data Fig. 1c, Supplementary Table 6 and Supplementary Fig. 2a,b). CLR-transformed cell type abundance revealed that cluster 4 and 6 CD14high classical monocyte abundances increase early during acute infarction. Simultaneously, cluster 0 CD4+ act 1 (CCR7low SOCS1high) T cell abundance and cluster 2 CD4+ act 2 (CCR7high SOCS1high) T cell abundance dropped during the disease course. In line with this, cluster 1 CD8+ T cell showed altered abundance across the immune response to ACS (Fig. 1e and Extended Data Fig. 1c).
Factor analysis extracts signatures that explain inter-patient variance
To unlock the full potential of our multiomic dataset from the Munich cohort and to identify overarching immune signatures, we preprocessed, integrated and harmonized data across the different data types (Methods and Supplementary Fig. 3a). The complexity of the integrated data made the analysis of the dataset with standard methods impractical. Therefore, we hypothesized that integrative factor analysis could exploit inter-patient variability to discover distinct immune signatures (that is, multicellular programs13) potentially allowing deduction of mechanistic and clinically relevant insights. MOFA enables the identification of major axes of variance including coordinated immune responses11 in a complex dataset that are not necessarily captured by individual features alone. Moreover, MOFA allows data integration across multiple data types (views). Here views represent (1) clinical laboratory markers, (2) cell-type-specific scRNA-seq-defined gene expression profiles, (3) circulating cytokines, (4) plasma-proteomics analysis and (5) neutrophil prime-seq14 data. MOFA extracted shared as well as view-specific combinations of features that describe variations of the circulating immune response (Fig. 1a and Supplementary Fig. 3a). Variance decomposition confirmed that the inferred MOFA factors indeed capture patterns spanning across different views (Fig. 1f and Supplementary Table 7). Furthermore, we analyzed the factor values at sample level to identify whether MOFA factors capture patterns that distinguish the different disease entities and timepoints (Supplementary Fig. 3b). This revealed several factors with diagnostic and mechanistic implications.
Identifying the superordinate immune signature during MI
MOFA identified factors that align to clinically relevant patterns: Factor 2 captured a large extent of inter-patient variance across the different views and explained most of the inter-patient variance in the clinical view (Fig. 2a and Supplementary Table 7). Factor 2 correlated with the development and resolution of myocardial ischemia reflected by the time course of clinical markers of myocardial damage and accurately discriminated CCS from ACS in patients with ACS with acute symptom onset and treatment without infectious complications (Figs. 1c and 2b). We therefore termed Factor 2 as ‘integrative ACS ischemia (IAI)’.

a, Overview of IAI (Factor 2). For each view, the heatmap shows the percentage of the variance that is explained by the factor. The bar plots show the total amount of features (left) and the relative amount of features (right; in respect to the number of view-specific features) among the top 1% of the highest-ranking features that influence the factor. The color coding on the left indicates the data type of each view: green, plasma proteomics; blue, neutrophil prime-seq; orange, cytokine measurements; dark orange, clinical values; purple, scRNA-seq data. The greyscale grading in the heatmap depicts the percentage of variance. b, IAI (Factor 2). Comparison of the factor values for each timepoint for sterile ACS, non-CCS and CCS. Mean ± s.e.m. values are shown. c, Replication of IAI in the Groningen cohort. Comparison of the factor values for each timepoint for ACS with controls. Mean ± s.e.m. values are shown. d, IAI (Factor 2). Normalized expression values of the top 0.5% of features for cluster 0 CD4+ T cell for sterile ACS and CCS. A longitudinal comparison of the normalized expression values (heatmap) and the weight of the features (bar plot) are shown. Plus and minus signs indicate the direction of the feature factor weight. e, Longitudinal comparison of normalized gene expression values of selected features for sterile ACS and CCS. For the following comparisons, only the post hoc test was significant: HINT1 Treg cells (cluster 11). Plus and minus signs indicate the direction of the feature factor weight. f, Phenotyping by flow cytometry of the effects of plasma obtained from ACS and CCS on monocytes isolated from healthy donors. Individual timepoints for sterile ACS are compared with those for CCS. Mean ± s.e.m. values are shown for mean fluorescence intensities (MFIs). No post hoc analysis was performed when the Kruskal–Wallis test was not significant. For b, c, e and f, parametric-distributed data were analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; nonparametric-distributed data were analyzed using the Kruskal–Wallis test with correction for multiple comparisons by Dunn’s test. In the case where only the ordinary one-way ANOVA or Kruskal–Wallis test, but not the multiple comparison, was significant, graphs are marked with a vertical bar on top. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
Next, we sought to replicate the temporal pattern of the immune response captured by IAI in the independent Groningen cohort (Fig. 1a). After matching the cell-type annotations of the two cohorts by applying the automated Groningen cell-type annotation strategy on the Munich data (Supplementary Fig. 4) and applying the Munich preprocessing and normalization approach in the Groningen cohort, we computed IAI by applying the feature factor weights identified in the Munich cohort (Supplementary Figs. 5 and 6) on cell-type-specific expression data in the Groningen cohort (Methods). IAI in the Groningen cohort indeed replicated the same time pattern (Fig. 2c).
Clinical markers of myocardial damage (troponin T, CK and CK-MB) had high factor weights on IAI, but these factor weights were lower than those of many omic variables (Fig. 2a, Extended Data Fig. 2 and Supplementary Table 8). In addition, the temporal pattern of IAI was reproduced after re-running MOFA without any clinical variables and the resulting factors did not change substantially (Supplementary Figs. 7 and 8), confirming that even though IAI captures a high amount of variance within the clinical markers and resembles the pattern of those, other multiomic views are important to define IAI.
Next, we dissected the molecular features contributing the highest factor weights to IAI. Cluster 0, 1 and 2 T cells and monocyte cluster 4 had the largest relative amount of highly weighted features across views (Fig. 2a). The expression of EIF3E and HINT1 in T cells (cluster 0) were among the highest-ranking features (Fig. 2d). In line with this, the gene product of EIF3E is required for robust T cell activation and regulates a burst in T cell receptor signaling15. HINT1 complexed with HSP70 has been shown to hold strong immunomodulatory functions in NK cells16. HMGB1 across T cells similarly held a high factor weight in IAI. HMGB1 promotes expansion and activation of T cells17. The normalized expression of these genes showed a comparable temporal development as IAI (Fig. 2d,e, Extended Data Fig. 2 and Supplementary Table 8).
Interestingly, T cell and monocyte clusters 0, 1 and 4, which contribute most of the features that constitute IAI, also showed significant alterations in cell type abundances across the disease course (Fig. 1e). CD4+ act 1 (CCR7low SOCS1high) T cells (cluster 0) were defined as activated CD4+ T cells as described before18. Monocyte cluster 4 showed an IL-1βhigh CD14high classical, inflammatory phenotype (Fig. 1d and Supplementary Fig. 2a,b). In line with this, incubation of monocytes with ACS plasma induced a proinflammatory phenotype with increased expression of effector surface molecules (CD93, CD88, CD14, CD86, CD89, CD11a) across ACS and downregulation of CD16 in early ACS. Interestingly, monocyte CCR2 dropped gradually across later stages of ACS plasma incubation (Fig. 2f and Extended Data Fig. 3a). Functionally, this was accompanied by a higher efferocytotic capacity of monocytes in vitro, without any changes in survival or reactive oxygen species (ROS) production induced by ACS plasma (Extended Data Fig. 3b).
Importantly, sub-analyses revealed that IAI was not influenced by preexisting medication, and in vitro co-incubation of human peripheral blood mononuclear cells (PBMCs) with heparin and platelet inhibitors (acetylsalicylic acid and prasugrel) did not reproduce the shifts observed in IAI, indicating that guideline-directed pharmacotherapy did not define the observed shifts (Extended Data Fig. 4 and Supplementary Fig. 9). Hence, IAI reflects the longitudinal pattern of myocardial ischemia and integrates clinical markers of myocardial damage with comprehensive multiomic information, summarizing the immune response to MI.
Distinct signaling pathways in monocytes characterize IAI
To allow for a more systematic understanding of characteristic immune signatures in IAI beyond the weight of individual features, we next sought to aggregate factor weights on the level of pathways. Across all our data views, several immune system pathways of the REACTOME19 and KEGG20 database were enriched (Supplementary Tables 9 and 10), including the interleukin (IL)-6, IL-10, IL-12 and IL-27 signaling pathways (Fig. 3a,b). The enrichment of the IL-6 cytokine signaling pathway was driven by high factor weights of IL6ST, STAT3 and JAK1 and SOCS3 in cluster 2 and 5 T cells and cluster 4 monocytes and plasma IL-6 (Fig. 3b–d, Supplementary Fig. 10 and Supplementary Table 8). In line with this, the inhibition of the intracellular IL-6 signaling cascade in monocytes activated ex vivo resulted in reduced effector functions such as ROS production, survival, efferocytosis, chemotaxis and proinflammatory cytokine secretion (Fig. 3e and Extended Data Fig. 5). This indicates that specific cell–cell or plasma–cell cytokine signaling influences immune shifts in ACS.

a, Positively enriched REACTOME immune system pathways in IAI (Factor 2) across all data dimensions for which at least 50% of genes have been included within the feature set. False discovery rate-adjusted P < 0.05. Coverage indicates the percentage of genes of the pathway that have been included in the analysis. Greyscale depicting P values. b, Factor weights of features (top 25%) in IAI (Factor 2) belonging to enriched interleukin pathways averaged across all views in the upper part of the heatmap (‘Pathway’) and shown per view in the lower part of the heatmap (‘View’). Heatmap depicts factor values of pathway genes. c, Normalized expression values of genes and cytokines belonging to the interleukin-6 signaling pathway for sterile ACS (clusters 0, 2 and 5) and CCS. A longitudinal comparison for CD4+ T cell clusters (clusters 0, 2 and 5) and cytokine features is shown. In cases in which only the ordinary one-way ANOVA or Kruskal–Wallis test, but not the multiple comparison, was significant, graphs are marked with a vertical bar on top. Plus and minus signs indicate the direction of the feature factor weight. d, Normalized expression values of genes belonging to the interleukin-6 signaling pathway for sterile ACS and CCS. A longitudinal comparison for CD14high monocyte genes (clusters 4 and 7) is shown (plus and minus signs indicate the direction of the feature factor weight). In c and d, parametric-distributed data were analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; nonparametric-distributed data were analyzed using the Kruskal–Wallis test with correction for multiple comparisons by Dunn’s test. e, Efferocytosis (n = 7), survival (n = 8), ROS production (n = 12) and chemotaxis (n = 7) of monocytes activated ex vivo with and without IL6ST inhibition. Comparison between control and treatment with the IL6ST-inhibitor group (paired dataset). Parametric data were analyzed using paired t-test (two sided); nonparametric data were tested using the Wilcoxon test (two sided). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Data points of paired data are connected by a line. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
Hence, we next hypothesized that cell–cell communication and cytokine–cell communication pathways might account for the observed immune states across patients and timepoints. We therefore focused on identifying the underlying ligand–receptor interactions between immune cells and the downstream signaling of receptors using the NicheNet model21. We selected only features with the highest factor weights to define candidate target genes and investigated ligands that showed a high regulatory potential and correlation of expression levels across samples with those target genes (Methods). Among the circulating cytokine features, this ligand–target analysis identified levels of circulating IL-6 to associate with expression levels of proto-oncogene Pim-1 in cluster 4 monocytes (Fig. 4a–e). High IL-6 levels also correlated negatively with monocyte CD74 expression, involved in antigen presentation22, but were accompanied by increased VCAN expression in cluster 7 monocytes (Fig. 4b–e). As we had observed a profound impact of IL-6 signaling on monocyte function and as a classical inflammatory phenotype was induced by ACS plasma, we analyzed the relevance of plasma IL-6 on influencing this phenotype in ACS (Figs. 2f and 3e, and Extended Data Figs. 3a and 5). Indeed, the activated monocyte phenotype induced by ACS plasma could be partially reversed by IL-6 inhibition across different timepoints, whereas IL-6 inhibition upon CCS plasma induction showed no difference on monocyte phenotype (Fig. 4f and Extended Data Fig. 6a).

a,b, Spearman correlations (|cor| ≥ 0.4) between ligand and target genes across all samples (n = 128). Target genes are selected as belonging to the top 1% of features with positive (a) or negative (b) feature weight in IAI (Factor 2). Ligands were selected based on a minimum regulatory potential score of 0.0012 for the shown targets according to the NicheNet model (corresponding to the 97% quantile of the regulatory potential score). Interactions mentioned in the main text are highlighted with a darker color. c, Spearman correlation scores (Cor) of selected ligand–target pairs from a and b. d, A longitudinal comparison of normalized expression values of selected genes for sterile ACS and CCS. Parametric-distributed data were analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; nonparametric-distributed data were analyzed using the Kruskal–Wallis test with correction for multiple comparisons by Dunn’s test. Plus and minus signs indicate the direction of the feature factor weight. e, Factor weights of the top 15 ligands with the highest factor weight in IAI (Factor 2). f, Monocyte phenotyping by flow cytometry after incubation with sterile ACS (TP4M) plasma with anti-IL-6 antibody or isotype control. All MFIs were normalized to the respective CCS plasma incubation average of the marker. Paired plasma incubation data with isotype (green dot) and IL-6 (red dot) inhibition are shown (TP4M n = 7). Parametric data were analyzed using the multiple paired t-test (two sided). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Data points of paired data are connected by a line. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
Specialized acquired T cell programs drive IAI
Besides IL-6, HMGB1, the gene encoding a DAMP that also showed a high weight on IAI, was involved in a plethora of correlated ligand–target pairs. In detail, expression levels of the ligand encoded by the gene HMGB1 in resting as well as activated T cell clusters 0, 1, 2 and 5 correlated with expression levels of most top-ranking target genes of IAI (Figs. 2d and 4a–c). HMGB1 might be related to the reduction of proteasome activity within the receiver cell as it showed a negative correlation with expression of UBC in cluster 0, 1 and 2 T cells, suggesting a reduced ubiquitination potential23. Similarly, expression of HMGB1 in activated cluster 0 and cluster 2 T cells correlated negatively with expression of PSME2 in cluster 4, 6 and 7 monocytes (Figs. 2d and 4c–e). Besides that, TGFB1 expression in cluster 1 T cells positively correlated with expression of ODC1 in cluster 4 monocytes, the gene product of which is known to inhibit inflammatory macrophage programs and macrophage apoptosis and induce efferocytosis24,25. ODC1 in these monocytes was downregulated early during ACS—however, it increased over time, suggesting an unleashed inflammatory monocyte state in early ACS (Fig. 4b–d).
Of note, cluster 2 T cell SELL (gene product: CD62L) was among the top-ranking ligands (Fig. 4e), which has been associated with a naive phenotype26—unexpected for an inflammatory response. However, a triphasic expression pattern of CD62L after T cell activation has been described, beginning with a fast shedding of CD62L (at the protein level) with subsequent re-expression within 24–48 h and finally a transcriptomic silencing27. Hence, the silencing of SELL at the transcriptomic level might not yet have occurred in early ACS. In line with this, the incubation of healthy PBMCs with ACS plasma from early timepoints resulted in an increase in CCR7− CD45RO+ effector memory T cells, indicating that ACS plasma at hyperacute timepoints initiates T cell maturation and activation, whereas ACS plasma from late timepoints showed a rather inhibitory effect on T cell activation (Extended Data Fig. 6b). Short-term overexpression of PD1 (ref. 28) and TIM3 (ref. 29) has been associated with T cell activation. In this setting, reduced expression after a short-term increase suggests subsequent inactivation. In summary, an early T cell maturation and activation state together with enhanced IL-6-dominated plasma cytokine levels might fuel an inflammatory monocyte phenotype and shape the immune landscape in ACS.
In addition to investigating synchronized changes occurring at the same timepoint, we also investigated lagged responses by correlating target gene expression to previous timepoint (lagged) ligand exposure (Methods). First, we investigated top-ranking ligands on IAI: this revealed that IL-6 correlated with CRP levels at later timepoints (Extended Data Fig. 7a–c), confirming the robustness of our analysis. IL-6 levels also correlated with multiple delayed acute-phase proteins (that is, ORM1, SAA2, HP and SERPINA3). Cellular HMGB1 from cluster 0, 1 and 2 T cells negatively associated with the expression of multiple T cell, monocyte and NK cell target genes (Extended Data Fig. 7b). Upon investigation of the top-ranking Factor 2 target genes, the ligands IL-6, IL-10, IL-12A and IL-27A, but also CXCL12 and CX3CL1, were found to positively associate with target genes of not only T cell phenotypes but also monocyte or NK cell phenotypes at later timepoints (Extended Data Fig. 7a). In general, multiple other cytokines including CXCL9 and CXCL10, which negatively influenced Factor 4 (described below), exhibited individual changes during ACS (Extended Data Fig. 7d). These data suggest a multilayered and complex immune trajectory to myocardial ischemia and reperfusion.
Factor analysis identifies an immune-repair signature in ACS: integrative ACS repair
We next tested whether our immunological MOFA approach could enable the detection and prediction of differences in the time from symptom onset to hospital presentation (that is, time of myocardial ischemia) as well as different clinical courses of MI (that is, recovery of myocardial function). IAI did not show significant differences between the subgroups of ACS (sterile ACS, ACS with delayed recanalization and ACS with hospital-acquired infection), with the latter depicting higher CK values (Fig. 5a–c). Yet, mechanistic and translational studies suggest a differential impact of immune states on myocardial healing and function30.

a, IAI (Factor 2). Longitudinal comparison of factor values in ACS subtypes (sterile ACS, ACS with hospital-acquired infection, ACS with delayed recanalization after vessel occlusion). b, Comparison of CK, CK-MB and troponin at early timepoints (TP1 and TP2) of ACS subtypes. c, Comparison of CRP levels between patients with sterile ACS and patients with ACS with hospital-acquired infection. d, Comparison of good and poor outcomes during hospitalization using EF levels. e,f, Longitudinal comparison of IAR factor (Factor 4) values (e) and CK levels (f) between patients with good and poor outcomes. g, ROC AUC plot for prediction of good versus poor outcomes for IAR (Factor 4), GRACE score, normalized CK levels, normalized CRP levels and normalized troponin levels at TP1M. h, ROC AUC plot for validation of prediction of good versus poor outcomes in the Groningen cohort. A lasso model was trained on the top features of IAR at TP1M and applied to the Groningen cohort (TP1G good outcome and poor outcome). i, Overview of IAR (Factor 4): for each view, the heatmap shows the percentage of the variance that is explained by the factor. The bar plots show the total amount of features (left) and the relative amount of features (right, in respect to the number of view-specific features) among the top 1% of the highest-ranking features that influence the factor. The color coding on the left indicates the data type of each view: green, plasma proteomics; blue, neutrophil prime-seq; orange, cytokine measurements; dark orange, clinical values; purple, scRNA-seq data. The greyscale grading in the heatmap depicts the percentage of variance. j, Factor values of the top 1% of features in IAR (Factor 4), showing only features belonging to clinical values (indicated by an asterisk), cluster 3 NK cell, cytokines and plasma proteomics. k, Normalized expression values of selected top features from NK cells in the Munich and Groningen cohorts, comparing patients with good and poor outcomes at TP1. The minus sign indicates the direction of the feature factor weight. l, ROC AUC plot showing the prediction results of a logistic regression model using selected NK features (CD53, GZMB, TXNIP) trained on the Munich dataset and applied to the Groningen dataset. For a–c and f, the data were analyzed using mixed-effects analysis with correction for multiple comparisons by Tukey’s test (a and b) or Šidák’s test (c and f). For d, e and k, the parametric datasets were analyzed using an unpaired t-test (two sided). For a–f, mean ± s.e.m. values are shown. *P ≤ 0.05; **P ≤ 0.01. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
To test whether distinct factors are associated with short-term clinical outcome, we selected patients with acute-symptom-onset ACS. The cohort was divided into two outcome groups: patients who showed a drop in ejection fraction (EF) during hospital stay were considered to have a poor outcome whereas patients with a favorable development of EF were considered to have a good outcome (Fig. 5d). We hypothesized that these variable disease courses might be associated with and/or influenced by different immune signatures. Indeed, MOFA Factor 4 had particularly low levels already at the time of hospital admission in patients with a poor outcome compared with levels in patients with improved cardiac function (Fig. 5e). We evaluated the potential of Factor 4 to distinguish between outcome at TP1M using the area under the curve (AUC) of the receiver operating characteristic (ROC) curve. Here Factor 4 levels showed better association with the development of EF than the Global Registry of Acute Coronary Events (GRACE) score31 and clinical markers at TP1M (Fig. 5f,g, Supplementary Fig. 11 and Supplementary Tables 11 and 12). In synopsis with its temporal development (enhanced after the acute ischemia and reperfusion phase), we termed this factor integrative ACS repair (IAR).
To corroborate the predictive value of the top-ranking features included in IAR already at the earliest timepoint and to reduce the number of relevant features, we trained a penalized logistic regression model that predicts outcome from these features at TP1M on the Munich cohort. We then applied this model on the Groningen cohort at TP1G. Again, the cohort was divided into two outcome groups (Methods). A comparison of the predicted and observed outcomes resulted in a ROC AUC of 0.83 on the replication cohort (Fig. 5h and Supplementary Tables 11 and 12), thus showing the ability of the model to generalize beyond the Munich cohort. This indicates the utility of novel integrative factors for outcome prediction and emphasizes the functional consequences of the immune response to myocardial recovery.
Distinct NK-cell- and plasma-derived immune signatures define IAR
Next, we investigated which features define IAR. Numerous features, mainly genes involving NK cell activation and cytotoxicity, such as TXNIP32, PRF133, LITAF34, GZMB33, FYN35, CST736 and CD5337, showed high negative weights on IAR (Fig. 5i,j and Extended Data Fig. 8). Moreover, multiple circulating cytokines strongly negatively influenced IAR: TRAIL (TNFS10) and CXCL9 showed high negative feature weights with a temporal development, which contrasted with IAR. CXCL10, also showing a high negative factor weight on IAR, has been associated with an unleashed recruitment of proinflammatory and cytotoxic T cells, making it a potential immune-pharmacological target molecule38. CCL24, known to adversely influence inflammation and fibrosis in several organs and to induce an M2 macrophage polarization39,40, also negatively influenced the outcome factor (Fig. 5j, Extended Data Fig. 8 and Supplementary Fig. 12). However, the plasma proteomics view revealed that circulating anti-trypsin enzymes of the SERPIN family (SERPINA1, SERPINA2, SERPINA3), which protect cells from granzyme-mediated cytotoxicity41, showed high positive factor weights on IAR and hence were associated with a more protective immune state. Interestingly, general markers of inflammation (that is, CRP, SAA1, SAA2 and C9 levels) were associated with an ongoing repair signature in this setting (importantly, patients with signs of relevant systemic inflammation at admission were not included in the analysis) (Fig. 5j). However, the isolated interpretation of single features within factors requires caution—a high rank does not necessarily correspond to the exact same biological behavior as the complete factor. Further prospective and mechanistic follow-up studies of top-ranking individual features will unravel their specific role in CS.
Investigating the potential of single features to distinguish outcome, we next asked whether a reduced set of NK cell markers and effector molecules (CD53, GZMB and TXNIP), holding high positive factor values, allows early separation by outcome. Distinct single features did not significantly differ between good- and poor-outcome groups at TP1M and TP1G, emphasizing the added value of composite signatures of multiple coordinated features to define IAR in its essence (Fig. 5k and Supplementary Fig. 12). Indeed, a logistic regression model including a combination of these few highly ranked features trained on the Munich data yielded an ROC AUC value of 0.79 on the Munich cohort as a training dataset and an ROC AUC of 0.91 in the Groningen validation cohort (Fig. 5l and Supplementary Tables 11 and 12). This highlights the potential of identifying a small subset of predictors among top-ranking IAR features that might serve as targeted clinical biomarkers for the prediction of myocardial recovery.
IC outlines the landscape of chronic coronary artery disease
Having characterized MOFA factors in ACS, we investigated which MOFA factor described patients with CCS compared with patients in whom CAD was securely ruled out (non-CCS). Factor 1 showed high positive values in patients with CCS, whereas patients with coronary sclerosis showed intermediate values. Patients with healthy coronary arteries showed mainly negative values of Factor 1 (Fig. 6a). We hence termed this factor integrative CCS (IC).

a, IC (Factor 1). Comparison of factor values for patients with CCS, coronary sclerosis (non-CCS) and healthy coronaries (non-CCS). Mean ± s.e.m. values are shown. b, Overview of IC (Factor 1). For each view, the heatmap shows the percentage of the variance that is explained by the factor. The bar plots show the total amount of features (left) and relative amount of features (right; in respect to the number of view-specific features) among the top 1% of highest-ranking features for the factor. The color coding on the left indicates the data type of each view: green, plasma proteomics; blue, neutrophil prime-seq; orange, cytokine measurements; dark orange, clinical values; purple, scRNA-seq data. The greyscale grading in the heatmap depicts the percentage of variance. c, Factor values of the top 1% of features showing only features belonging to CD4+ T cells (clusters 0 and 2) in IC (Factor 1). d, Normalized expression values of selected top features of IC (Factor 1) for samples classified as CCS and non-CCS (plus or minus signs indicate the direction of the feature factor weight). e, Factor weights of the top 10 ligands with the highest factor weight in IC (Factor 1; plus or minus signs indicate the direction of the feature factor weight). f, Normalized expression values of selected top ligands of IC (Factor 1) for samples classified as CCS and non-CCS (plus or minus signs indicate the direction of the feature factor weight). For a, Parametric-distributed data were analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Tukey’s test. For d and f, parametric data were analyzed using an unpaired t-test (two sided) and nonparametric data were tested using the Mann–Whitney test (two sided). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Exact P and n values are summarized in Supplementary Tables 13 and 14, respectively.
Next, we again analyzed which features dominate IC: the factor was defined by many features of CD4+ and CD8+ T cells and captured mainly the variance of those views (Fig. 6b). Expression of modulators of T cell antigen recognition, signal transduction and T cell activation such as gene products of CD3E, ICAM3 and TRAC42,43 in activated cluster 0 T cells had a high positive weight on IC. In line with this, expression of PRDX2, CORO1A, JUNB and CD37 showed strong positive associations with IC. FOSB expression in activated cluster 0 T cells, cluster 1 T cells and cluster 3 NK cells had a negative weight on IC (Fig. 6c,d, Extended Data Fig. 9a and Supplementary Table 8).
We further explored ligand–target interactions within IC21. Interestingly, ICAM3 expression in several clusters was among the top 10 ligands with the highest weights on the factor. Simultaneously, gene expression of the gene product ligands CALM1 and CALM2 in similar clusters showed negative factor weights on IC (Fig. 6e). ICAM3 or CALM1 from cluster 0 and 11 T cells negatively correlated with PTMA expression in cluster 1 T cells. Moreover, CALM1 in multiple CD4+ T cell as well as B cell and monocyte clusters was associated with PTMA expression in cluster 1 CD8+ T cells (Fig. 6e,f and Extended Data Fig. 10), which holds broad disease-protective effects44.
What sustains the CD4+ T cell phenotype in IC? NAMPT in cluster 9 and 12 monocytes (negatively associating with IC) showed a negative correlation with JUNB and other phenotype-defining genes in T cell clusters (Extended Data Fig. 10b, Supplementary Fig. 13a,b and Supplementary Table 8). NAMPT has been described to be a key regulator of monocyte differentiation particularly during inflammatory states45 and has been described to be increased in patients with ACS as well as in M1 inflammatory macrophages46. In summary, the IC signature was associated with CCS and was defined by a dysregulated activation pattern of the monocyte and T cell compartment.
Discussion
The systemic immune signatures of ACS and CCS remain incompletely understood in humans, but are highly relevant for atherosclerosis, thrombosis and myocardial remodeling. In particular, insights from human multiomics studies might provide leads for developing new strategies for accessible biomarker signatures for diagnosis and prognosis as well as tailored anti-inflammatory therapies in ACS and CCS.
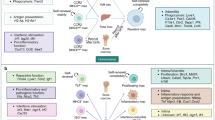
In line with this, IAI captured the immune response in ACS in two independent cohorts. It was mainly defined by features derived from CD4+ and CD8+ T cells and monocytes and multiple plasma–cell and cell–cell communication pathways: ACS plasma induced a functional and phenotypic proinflammatory shift in monocytes, which could be partially reversed by inhibition of IL-6 signaling. Similarly, T cell activation with subsequent quiescence was induced using ACS patient plasma depending on the timepoint. Multiple T-cell-derived ligands, such as TGFB1 and HMGB1, defined the communication with monocytes and were associated with expression of downstream targets in monocytes. In line with this, distinct CD4+ T cell phenotypes have been associated with atheroprogression or plaque rupture in CS47,48,49,50. In the OPTICO-ACS study, flow cytometric analysis emphasized an important role for T-cell-derived cytotoxic effector molecules in plaque-erosion ACS49.
Although all MOFA factors were estimated in an unsupervised manner, using neither explicit information on outcome nor the trajectories of specific patients, IAR was associated with the functional treatment outcome of patients with STEMI already at hospital admission. This finding enabled the training of a supervised model that could then be applied to a second independent cohort, providing evidence for the robustness and generalizability of the IAR factor. IAR was defined by negatively high-ranking features in NK cells such as TXNIP32, GZMB33 and CD5337, the gene products of which implicate NK cell cytotoxicity as a possible predictive marker of adverse outcome. Indeed, previous studies on lymphocyte-mediated cytotoxicity showed a deleterious role on post-ischemic cardiac remodeling, atherosclerosis and pulmonary vascular permeability51,52,53.
IC, again estimated in an unsupervised way, showed higher levels in patients with CCS than in patients without CCS, but this needs further validation in larger cohorts. High-ranking features contributing to IC indicated a dysfunctional T cell phenotype, possibly induced by a disrupted CD16high monocyte–NAMPT–T cell signaling axis.
Our study holds multiple important implications for clinical, basic and translational cardiovascular research: first, we provide proof-of-concept evidence that multiomic profiling of the circulating immune signature paired with MOFA has the potential to estimate disease state, phenotype and outcome of remote, non-accessible injury sites (that is, myocardium). By reducing the very large number of potential predictors to few factors, we identify overarching signatures of clinical significance. This implies that (1) integration and factor-based analysis of complex datasets can add to the crucial understanding of (cardiovascular) disease and (2) multiomic liquid biopsies without access to the tissue site of injury offer a promising concept for future biomarker studies. Small sets of top-ranking features (identified from large-scale clinical MOFA studies) could serve to identify patients at risk and possibly trigger early initiation of heart failure treatment, more extensive revascularization or prolonged intensive care monitoring54.
From a pathophysiological perspective, our data highlight distinct plasma–cell- and cell–cell-imprinted, monocyte-, NK-cell- and T-cell-dominated trajectories over the disease course. In monocytes, these associate with a proinflammatory phenotype and function that can partially be ameliorated by inhibition of plasma-mediated IL-6 signaling. In T cells, early ACS plasma induced activation and later ACS plasma induced deactivation. Crucially, a specific signature was characterized by reduced NK cell cytotoxicity and enhanced influence of circulating anti-trypsin SERPINs (known to protect from cytotoxicity41). This was associated with later repair phases of ACS and correlated with favorable short-term outcome. This underscores the possible functional importance of the identified axes and supports the concept of therapeutic immune modulation to limit cardiac damage.
However, this study also has limitations: it was designed as an observational proof-of-concept study allowing a comprehensive characterization of immune signatures in CS using MOFA. A restriction of multiomics studies, such as this, is the limited sample size that reduces the generalizability, yet—based on the fine-grained analyses—particularly provides hypotheses for future biomarker and mechanistic studies. The power is not comparable to a large-scale biomarker trial, which is indispensable to evaluate the ultimate diagnostic performance of MOFA factors and to probe translation into the clinical setting. The methods used in this paper are highly cost-intensive, but selected candidate biomarkers could be assayed in a targeted way for such larger clinical cohorts. The study focuses on STEMI patients without including other forms of ACS. Furthermore, patients in the ACS cohorts of the Munich and Groningen study received guideline-recommended treatment that potentially influenced the immune signatures. Yet, analyses of medication-specific effects on factor values suggest no relevant confounding.
In summary, this multiomics study of coronary syndromes identifies systemic immune states in CCS and ACS. MOFA, in combination with downstream analyses, reveals the underlying molecular, plasma–cell and cell–cell communication pathways that mechanistically translate to functional and phenotypic shifts in monocytes and T cells in vitro. This underlines MOFA as an innovative approach enabling the dissection of multicellular immune programs with mechanistic and clinical implications in cardiovascular disease. Moreover, our study, as a globally available resource, provides novel targets for experimental studies and large-scale MOFA-driven biomarker trials and informs prioritization of novel candidate targets for immune-modulatory interventions in CS.
Methods
Munich cohort: ethics and patient cohort
Munich cohort (M): Informed consent was obtained from the patients in accordance with the Declaration of Helsinki and with the approval of the Ethics Committee of LMU Munich (number 19-274). We collected blood from n = 62 patients using repetitive serial sampling and separately analyzed the different immune cell constituents. For blood collection, we used heparin-anticoagulated blood (Sarstedt, catalog number 02.1065.001). Blood processing was performed within 2 h of collection on average across the Munich and Groningen cohorts. A total of n = 125 whole blood tests, n = 122 PBMC samples, n = 246 plasma samples and n = 121 polymorphonuclear neutrophil (PMN) samples were used for analyses. In the ACS group, patients with STEMI were included and blood was analyzed longitudinally. Blood sampling was done peri-interventionally (TP1M)—during catheterization to avoid time loss, 14 h (±8 h) after intervention (TP2M), 60 h (±12 h) after the acute event (TP3M) and before discharge, about 5–8 days after the acute event (TP4M). The patients were further subdivided into those without direct reperfusion within 24 h after symptom onset (delayed myocardial reperfusion, n = 4) and patients with direct reperfusion within 24 h due to coronary intervention (acute MI, n = 24). A subgroup of patients with evidence of infection in laboratory testing who were treated with antibiotics in the clinical setting defined a subgroup with hospital-acquired infection (n = 5), which was differentiated from the sterile group with STEMI and without hospital-acquired infections (n = 19). The latter was used for comparison with the CCS group. Patients were also subdivided based on clinical outcome. For this purpose, the EF measurement was determined according to Simpson’s method in echocardiography. A comparison was made between the findings on admission and during the hospital stay (first EF value) or before discharge (second EF value) (resulting in a ΔEF). Based on these, a classification was made according to positive and stable (good outcome) and negative (poor outcome) ΔEF in the acute setting.
The CCS group included patients with an initial diagnosis of CCS based on a cardiac catheterization (lumen reduction of >50%) or coronary CT scan (>75th percentile; CCS, TP0M n = 16). Coronary healthy patients, with CAD ruled out by catheterization or CT, were included as a comparison group for the CCS group (non-CCS, TP0M n = 18). Coronary sclerosis was defined as coronary irregularities without substantial lumen obstruction (<50%).
Exclusion criteria for the Munich cohort were cardiogenic shock, age >85 years and <30 years, severe systemic diseases (chronic liver disease, active hemato-oncologic diseases, active cancer, autoimmune diseases, acute inflammatory event with a fever or CRP > 2 mg dl−1 at admission (except for patients with a subacute ACS with delayed recanalization who regularly showed elevated CRP levels as an already ongoing delayed response to the infarction) and the use of immunosuppressants at inclusion. For the CCS cohort, patients with relevant elevation of troponin T levels were also excluded.
Analysis modalities for the Munich cohort (if not specified otherwise)
Clinical blood test
The clinical blood tests were performed as part of the treatment during hospitalization. We involved the following clinical biomarkers and blood cells: CK, CK-MB, troponin T, CRP, leukocytes and neutrophils.
Cytokine and chemokine assays
For the isolation of plasma, 2 × 1 ml whole blood was centrifuged at 2,000 × g (relative centrifugal force) for 20 min at 4 °C (Centrifuge 5424 R, Eppendorf AG). Afterward, the supernatant was carefully removed and pooled in a common Eppendorf reaction vessel for cryopreservation at −80 °C.
For detection and quantitation of cytokines and chemokines, samples were sent on dry ice to EveTechnologies where a Human Cytokine/Chemokine 71-Plex Discovery Assay array (HD71) was performed. Within the assay, the following biomarkers were determined: 6CKine, BCA-1, CTACK, EGF, ENA-78, eotaxin, eotaxin-2, eotaxin-3, FGF-2, Flt3L, fractalkine, G-CSF, GM-CSF, GROα, I-309, IFNα2, IFNγ, IL-1α, IL-1β, IL-1RA, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-15, IL-16, IL-17A, IL-17E/IL-25, IL-17F, IL-18, IL-20, IL-21, IL-22, IL-23, IL-27, IL-28, IL-33, IP-10, LIF, MCP-1, MCP-2, MCP-3, MCP-4, M-CSF, MDC, MIG, MIP-1α, MIP-1β, MIP-1δ, PDGF-AA, PDGF-AB/BB, RANTES, sCD40L, SCF, SDF-1α + β, TARC, TGFα, TNFα, TNFβ, TPO, TRAIL, TSLP and VEGF-A.
Plasma proteome analysis
The isolation and storage of the plasma were mentioned above. The plasma in vials were slowly thawed at +4 °C and mixed at a ratio of 1:5 with a proteomic buffer (2% SDS (Thermo Scientific, catalog number J22638.AE), 2.5 mM DTT (Invitrogen, catalog number P2325) in 50 mM Tris (Invitrogen, catalog number AM9820)). Afterward, the samples were immediately boiled at 95 °C for 10 min and cryoconserved at −80 °C. Plasma samples were prepared by SDS lysis, automated SP3 cleanup and tryptic digest (as also described before)55,56. Samples were measured on an Orbitrap Exploris 480 instrument (Thermo Fisher Scientific) in label-free data-independent acquisition mode while separating peptides on a 44 min gradient on a nanoEASY 1200 system (Thermo Fisher Scientific) coupled to the mass spectrometer. Raw files were analyzed in Spectronaut 14 (Biognosys57) against a spectral library that was generated from 52 fractions measured (as also described before55). A false discovery rate cut-off of 0.01 was applied, and spectra were searched against a human Uniprot database from 2018 including isoforms. For data filtering, the option Q value percentile with a fraction of 0.2 was used and global normalization using the median was applied. Further downstream analysis was performed in R. Normalized intensities were filtered for at least 80% valid values per row and column; remaining missing values were median centered and imputed using a randomized Gaussian distribution with a downshift of 1.8. Plasma proteomics was included into the MOFA to identify patterns of variance. No differential expression data were reported in the paper.
Isolation of PMNs
Initially, 400 µl of whole blood was added to a tube and 20 μl each of the Isolation Cocktail and RapidSpheres (EasySep Direct Human Neutrophil Isolation kit, STEMCELL Technologies, catalog number 19666) were added. After 5 min of incubation at room temperature, the reagent was filled up to 4 ml with PBS (Dulbecco’s phophate-buffered saline (1×), Thermo Fisher Scientific, catalog number 14190-094) + 1 mM ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich Chemie, catalog number 03690). Subsequently, the tube was placed in a magnet (EasySepMagnet, STEMCELL Technologies) for 5 min. Then, without being removed from the magnet, the contents of the tube were transferred to a new tube in a continuous motion. After 20 μl of RapidSpheres was added again, the incubation steps were repeated without PBS + 1 mM EDTA being added. After another decantation, the new tube was placed in the magnet for 5 min without the addition of RapidSpheres. The newly decanted tube was centrifuged at 320 × g for 7 min at 4 °C (Centrifuge 5810 R, Eppendorf AG). The pellet was then resuspended in 100 μl of PBS. Subsequently, 10 µl of the suspension was used to adjust the concentration to 5 million cells ml−1. For this, the dead cells were stained with trypan blue (Sigma-Aldrich Chemie, catalog number T8.154) and the concentration was calculated using a Neubauer counting chamber (LO-Laboroptik). For cryopreservation at −80 °C, cell suspension was added to RLT Plus buffer (Qiagen, catalog number 1053393) containing 1% 2-mercaptoethanol (Sigma-Aldrich Chemie, catalog number M3148) at a ratio of 1:10.
Prime-seq
For the analysis of the transcriptome of PMNs, prime-seq14, an early barcoding bulk RNA-seq method, was used. Samples were pretreated with proteinase K (Life Technologies, catalog number AM2548) followed by isolation with cleanup beads (Sigma-Aldrich, catalog number GE65152105050250; ratio, 2:1 beads per sample). DNase I (Thermo Fisher, catalog number EN0521) was used to digest the cells to make the transcriptome accessible for the process of reserve transcription. This was done by adding 30 units of Maxima H enzyme (Thermo Fisher, catalog number EP0753) and 1× Maxima H buffer (Thermo Fisher, catalog number EP0753), 1 mM dNTPs (Thermo Fisher, catalog number R0186), 1 µM template-switching oligo (Integrated DNA Technologies (IDT)) and 1 µM barcoded oligo-dT primers (IDT), and incubating for 90 min at 42 °C (reaction volume, 10 µl). After all samples were pooled, they were purified in a 1:1 ratio with cleanup beads. For the elimination of the leftover primers, exonuclease I (NEB, catalog number M0293L) was added (incubation setup: 37 °C for 20 min, then 80 °C for 10 min), followed by another purification with cleanup beads. The synthesis of the second strand of complementary DNA (cDNA) was prepared by adding 1× KAPA HotStart Ready Mix (Roche, catalog number 07958935001) and 0.6 µM SINGV6 primer (IDT) (reaction volume, 50 µl). For amplification, subsequent PCR cycles were performed: start: 98 °C for 3 min; 15 cycles: 98 °C for 15 s, 65 °C for 30 s and 72 °C for 4 min; end: 72 °C for 10 min. To repurify the sample, cleanup beads were added at a ratio of 0.8:1 beads per sample and dissolved out in 10 µl DNase-and-RNase-free distilled water (Thermo Fisher, catalog number 10977-049). Quantification and size selection of the purified cDNA were then performed using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher, catalog number P7581) and the High-Sensitivity DNA Kit (Agilent, catalog number 5067–4627). For library preparation, a fivefold-lower reaction volume of the NEBNextUltra II FS Library Preparation Kit (NEB, catalog number E6177S) than recommended by the manufacturer was used. Fragmentation of cDNA was performed using the enzyme mix and the reaction buffer (reaction volume, 6 µl), and ligation was performed using Ligation Enhancer, Ligation Master Mix and a custom prime-seq adapter (1.5 µM, IDT; reaction volume, 12.7 µl). Solid-phase reversible immobilization-select beads (Beckman Coulter, catalog number B23317) were then used for a double size selection (ratio of 0.5 and 0.7). For amplification, Q5 Master Mix (M0544L, NEB), 1 µl i7 Index Primer (Sigma-Aldrich) and 1 µl i5 Index Primer (IDT) were used followed by PCR (start: 98 °C for 30 s; 13 cycles: 98 °C for 10 s, 65 °C for 75 s and 65 °C for 5 min; end: 65 °C for 4 min). After successful size selection with solid-phase reversible immobilization-select beads and a quality check, the libraries were sequenced using NextSeq (Ilumina).
The sequencing reads were processed using zUMIs pipeline using the Gencode human release version (https://www.gencodegenes.org/human/release_35.html). Only barcodes matching the expected samples were considered and exported as count matrices, both raw counts and library-size normalized ones. First, the data were checked using fastqc (version 0.11.8 (ref. 58)). Regions on the 3′ end of the fragment reading into the poly-A tail were removed by Cutadapt (version 1.12 (ref. 59)). The zUMIs pipeline (version 2.9.4d)60 was applied, filtering the data, with a phred threshold of 20 for 4 bases the unique molecular identifier and barcode, mapping the reads to the human genome with the Gencode annotation (v35) using STAR (version 2.7.3a); reads were counted using RSubread (version 1.32.4)61,62.
PBMC isolation
For isolation of PBMCs, 8 ml of whole blood was transferred to a BD Vacutainer CPTTM (Becton Dickinson, catalog number 362780), swiveled twice and centrifuged at 1,650 × g for 20 min at room temperature (Centrifuge 5810 R, Eppendorf AG). After swiveling twice, the supernatant was transferred into a 15 ml tube and a further centrifugation step with 350 × g for 7 min at 4 °C was performed. The resulting cell pellet was resuspended in 4 ml freezing medium and aliquoted. The freezing medium consisted of 45% Roswell Park Memorial Institute (RPMI) medium (VLE-RPMI 1640, Bio&SELL, catalog number BS.52551528.5) with 1% glutamine (Gibco l-glutamine (200 mM), Thermo Fisher Scientific, catalog number BS.K0283), 45% fetal bovine serum (FBS; FBS SUPERIOR stabil, Bio&SELL, catalog number FBS.S0615) and 10% dimethyl sulfoxide (DMSO, Sigma-Aldrich Chemie, catalog number D2438). For cryopreservation, samples were slowly frozen in a Mr. Frosty freezing container (Thermo Fisher Scientific, catalog number 5100-0036) at −80 °C for 24 h and then transferred to −80 °C freezers.
FACS and scRNA-seq preparation
For scRNA-seq analysis of the frozen PBMCs, an adapted thawing protocol of 10× was used63. Samples were thawed at 37 °C for 3 min. This was followed by stepwise dilution (5 × 1:1) with dropwise addition of complete growth medium. The complete growth medium consisted of 10% FBS and 90% RPMI. The sample was then filtered using a 50 µm strainer and centrifuged at 300 × g for 5 min at room temperature. The supernatant was removed to the last milliliter, and the cell pellet was resuspended in it by using a wide-bore pipet. An additional 9 ml of complete growth medium was slowly added, and the sample was split into two. A further centrifugation step at 300 × g for 5 min at 4 °C was performed. One-half of the sample was used for further processing for scRNA-seq analysis. The cell pellet was resuspended in 100 µl Fc block (BD Pharmingen, catalog number 564200; 1:50) and incubated on ice for 10 min. To label the cells, TotalSeqB anti-human hashtag antibodies (1:500; BioLegend; Supplementary Table 15) were added to the sample and then incubated at 4 °C for 30 min. To maximize the performance, TotalSeqB anti-human hashtag antibodies were pre-centrifuged at 14,000 × g at 4 °C for 10 min. Following this, the sample was washed three times by adding 5 ml of FACS buffer (PBS with 0.5% BSA (Albumin Fraktion V, Carl Roth, catalog number 8076.4)) and centrifugation at 250 × g for 10 min at 4 °C each time. After the last centrifugation step, the cell pellet was resuspended in 0.04% BSA in PBS and the concentration was adjusted to 200 cells µl−1 using a Neubauer counting chamber. Lastly, marked samples were pooled. The other half of the sample was used to prepare the FACS analysis. The sample was incubated with 200 µl Fc block (1:50) at 4 °C for 10 min. Staining of the cells was done by a 20 min incubation with an antibody master mix (1:400; Supplementary Table 15). After centrifugation at 300 × g for 7 min at 4 °C, the cell pellet was resuspended in 300 µl FACS buffer. The dead cells were stained immediately before flow cytometry with an LSRFortessa Flow Cytometer (BD Biosciences). The flow cytometry data were analyzed with FlowJo (BD, version 10.8.14). Flow cytometry allowed the analysis of large cell counts. Rare populations such as dendritic cells were captured at representative counts, allowing reliable detection of differences. The scRNA-seq captured only a fraction of these cells but was suitable for the performance of unsupervised immune cell profiling of more frequent immune cell subsets, but not for the detection of differences in geometric averages of rare ones. Rather, scRNA-seq enabled an unsupervised sub-clustering of more common immune cells not identified by canonical surface-based flow cytometry panels due to limitations in the number of antibodies used. The CLR-transformed flow cytometry data were hence provided as a baseline characterization of the sub-cohorts. For the MOFA-model-based deep phenotyping, the gene expression profiles of the fine-grained scRNA-seq sub-clusters were included. The geometric averages of immune cell subsets were not included into the MOFA model but were used for an initial screening of differences in canonical immune cell subsets as a baseline characterization. Previous CLR transformation was performed as described in the scRNA-seq compositional analysis section.
scRNA library preparation and sequencing
For single-cell sequencing, libraries were prepared using the Chromium Next GEM Single Cell 3′ Reagent Kit v3.1 (CG000206 Rev D with with all mentioned components) from the 10x Genomics protocol. Barcode-based multiplexing with TotalSeqB anti-human hashtag antibodies (Supplementary Table 15) was performed to reduce artefacts associated with batch variation. According to the manufacturer’s instructions, the gel beads in emulsion were first prepared to obtain cDNA with reserve transcription. After purification of the cDNA, amplification and size selection were performed. After final quantification and quality control, the gene expression and cell surface libraries were constructed for sequencing. Sequencing was subsequently performed by IMGM Laboratories using Illumina NovaSeq 6000.
Bioinformatics analysis of the CS scRNA-seq dataset
Preprocessing
SC data preparation: cellranger
After sequencing, the FASTQ files for the gene and cell surface libraries were processed using the cellranger count pipeline (chemistry: Single Cell 3′ v3; pipeline version 3.1.0). Each sample was mapped to the human reference genome (GRCh38; version: 3.1.0). The library and reference files were created according to the 10x Genomics instructions and example files for Antibody Capture with TotalSeq B (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/feature-bc-analysis#feature-ref (17 January 2023)).
The pipeline quantified each feature (genes plus antibodies) in each cell and generated quality control summaries and feature barcode matrices for each of the 14 libraries (Supplementary Table 4).
For further analysis, we took the ‘filtered_feature_bc_matrix.h5’ of each library and split it up into two separate anndata objects: one containing the gene expression and one the antibody capture counts.
Demultiplexing and doublet identification
For the demultiplexing of the scRNA-seq data, we took the antibody capture count anndata objects of each library, converted them to Seurat objects, normalized the counts with CLR transformation and applied the ‘HTODemux’ function of the Seurat package (version: 4.1.1) as described in the vignette with the default 0.99 quantile threshold to classify the cells as positive or negative for each hashtag oligo. Cells that have been classified as positive for more than one hashtag oligo have been annotated as doublets.
Cell quality control and filtering
In a next step, we transferred the cell annotation results from the demultiplexing to the gene expression anndata files and applied some cell quality control criteria and filtering based on the gene expression counts to remove low-quality cells. These steps were done for each library separately.
In a first basic filtering step, we kept only cells that have counts on at least 200 genes and genes that have counts in at least 3 cells. Furthermore, we combined the percentage of mitochondrial gene count (pct_counts_mt), number of genes by count (n_genes_by_counts) and total count (total_counts) criteria to filter out further cells. We only kept cells that have:
-
n_genes_by_counts < 5,000 ∩ total_counts < 20,000
-
n_genes_by_counts > 500 ∩ pct_counts_mt < 15
Subsequently the data were normalized (10,000 counts per cell) and log transformed (log1p) using the scanpy toolkit64 1.8.1 in Python v.3.9.6. Furthermore, we excluded mitochondrial and ribosomal genes as they were not of interest for the analysis.
Data integration, clustering and cell-type annotation
To get a joint embedding of the complete dataset and correct for potential batch effects between the libraries, we took the processed data from the quality control and applied the Scanorama method (scanorama.correct_scanpy, v 1.7.1) using 2,000 highly variable genes, batch-size parameter of 2,000 and default parameters. This returned a Scanorama-corrected count matrix and a joint embedding. We compared the data integration before and after applying Scanorama by inspecting the uniform manifold approximation and projection (UMAP) plots before and after (Supplementary Fig. 14a–c) and by calculating the local inverse Simpson’s index (LISI) score (compute_lisi) with the corresponding function of the LISI R Package (https://github.com/immunogenomics/LISI).
Subsequently, we used the Scanorama embedding as input for the computation of a neighborhood graph (scanpy.pp.neighbors, n_neighbors=10, n_pcs=50) and the subsequent clustering of the cells using the Leiden algorithm (scanpy.tl.leiden; default parameters).
We found 18 different clusters that we annotated manually by looking at the expression patterns of PBMC marker genes selected based on literature research and calculating differentially expressed genes between the clusters using a Wilcoxon rank sum test with Benjamini–Hochberg adjustment as implemented in the scanpy framework (scanpy.tl.rank_genes_groups). With this strategy, we could annotate all 18 clusters to all common major peripheral blood mononuclear immune cell types (Supplementary Fig. 2a,b).
Compositional analysis
To investigate compositional changes of the cell-type clusters between the different patient groups and timepoints, we determined the percentage of cells that have been assigned to the different cell-type clusters for each patient and timepoint separately (for each patient and timepoint: amount of cells belonging to the cluster/total amount of cells) and adjusted them with CLR transformation. Adjusted values were then analyzed using ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test (*P ≤ 0.05; **P ≤ 0.01).
Multiomics data integration: MOFA
Data harmonization and integration
For the integrated and combined analysis of all the different data sources (single-cell data, cytokines, neutrophils, plasma proteomics and clinical values), we applied several preprocessing and normalization steps separately on the features of the different types of data to make them comparable and adjust the distributions.
CS scRNA-seq data
We applied the pseudo-bulk approach to summarize single-cell data on the level of cell-type (cluster) specific gene expression per sample because all other omics data were measured on the bulk level. To this end, we calculated for each of the identified 18 cell-type clusters for each sample (=patient and timepoint) the mean counts across all cells. Afterward, we adjusted the gene counts of each sample in each cluster with a scaling factor so that each sample has the same amount of counts across all genes to account for technical differences in sequencing depth between the samples.
To ensure that we only consider reliably expressed genes, we applied additional filtering steps on the genes and clusters:
We excluded clusters 14–18, which had only very low numbers of cells per patient and timepoint (mostly less than 10 cells).
We filtered out genes based on the total number and percentage of cells that expressed those genes in the corresponding cluster, keeping only genes that fulfill one of the criteria below:
-
Percentage of cells expressing gene > 50 ∩ total number of cells expressing gene > 1,200
-
Percentage of cells expressing gene > 40 ∩ total number of cells expressing gene > 3,000
The thresholds were chosen considering that genes should be detectable in a high number of cells and in several samples, but at the same time, a considerable number of genes for each cluster should be kept.
After filtering, we log transformed the count values and applied quantile normalization as further normalization steps to align the distributions of gene expression levels between the samples.
This resulted in 11,831 features (which correspond to genes) across all the different clusters (ranging from 315 for cluster 13 and 2,159 for cluster 4), which we used as input for the different cell-type cluster dimensions from the single-cell data for the MOFA (Supplementary Fig. 3a).
Cytokines
To prepare the cytokine data for integration with the other datasets we set ‘OOR’ values to 0 and log transformed the values to adjust the distributions after adding a pseudo-count of 1 to all values. Furthermore, we excluded cytokines which have valid measured values in less than 20% of the samples. In total this resulted in 65 different cytokines which have been used as input features for the integrated analysis (Supplementary Fig. 3a).
Neutrophils
As input features from the neutrophil dimension, we took the umi exon counts from the prime-seq and applied the processing steps below to align the reads with the scRNA-seq data, adjust for potential technical effects and strictly remove samples and genes with low-quality reads.
In a first step, we adjusted the gene names and mapped them from ‘ENSEMBL’ gene ids to ‘SYMBOL’ gene ids. Then, we filtered out ribosomal and mitochondrial genes as we also excluded them in the scRNA-seq data preprocessing, and they are not relevant for the analysis. Furthermore, we excluded genes that are not expressed in at least 80% of the samples and removed samples that do not have reads in at least 90% of the remaining genes. In the next step, we adjusted for differences in sequencing depth between the samples and normalized the counts with a scaling factor so that the sums of reads for each sample across all genes are the same. Then, we logarithmized the resulting counts. Finally, we decided to keep only highly variable genes, so we removed all genes whose variance lies below the 25% quantile of the variance distribution. This resulted in a total of 892 genes measured on 92 samples that are considered as input features for the neutrophil dimension. As for the scRNA-seq data, we applied quantile normalization to the counts in a final normalization step (Supplementary Fig. 3a).
Plasma proteomics
For proteomics, we used the same preprocessing and normalization steps as described in the previous ‘Plasma proteome analysis’ section and took the resulting normalized and median-centered intensities measured for 490 different proteins as input features of this dimension (Supplementary Fig. 3a).
Clinical values
As input features of the clinical data dimension, we used the measured CK, CRP, CK-MB and troponin values and log transformed them (Supplementary Fig. 3a).
Model training
After these individual preprocessing steps, we had in total 13,382 features across 18 different dimensions (referred to as views throughout the paper) resulting from (1) clinical values, (2) cell-type cluster 1–14 of the scRNA-seq data, (3) cytokines, (4) plasma proteomics and (5) neutrophils (Supplementary Fig. 3a). We applied feature-wise quantile normalization onto the quantiles of the standard normal distribution for all data types.
Then we trained the MOFA model using the R/Bioconductor package MOFA2 (version 1.2.2) with maxiter parameter 50,000 to ensure convergence and 20 factors and the remaining default parameters. The number of estimated factors was chosen to balance the trade-off between explained variance and low number of factors. Also, we tested the influence of the specified number of factors on the model results by running alternative MOFA models with 5, 10, 15 and 25 factors and comparing them with the 20-factor model. We found that especially the first five inferred factors are not substantially affected by the choice of the number of factors (Supplementary Figs. 3a and 14d and Supplementary Table 16).
To evaluate the effect of the clinical features, we trained a second MOFA model excluding the 4 clinical features with in total 13,378 features and compared the resulting factor values and feature weights to the original model (Supplementary Figs. 7 and 8a,b).
Downstream analyses
Gene set enrichment analysis: pathways
On the feature weight matrix resulting from our trained MOFA model, we conducted pathway enrichment analysis for the first five inferred factors of the MOFA model using the gene set annotations from the REACTOME19 and KEGG65 databases. We tested all pathways belonging to the ‘Immune System’ category in REACTOME (n = 191) and pathways that are classified as ‘Immune system’ or ‘Signal transduction’ pathways in KEGG (n = 52).
To test the enrichment of the pathways across all our data input views, we generated an extended pathway gene annotation set for those pathways in which a feature (consisting of data dimension, and gene and protein code) was considered to belong to the pathway if the gene and protein code maps to the genes annotated to the pathway. Features included in the MOFA but not within the pathway were taken as background set. To map the gene and protein codes to the gene-set annotations in KEGG and REACTOME (reacome.db, version 1.76.0; ReactomePA version 1.36.0), we used the bitr function from the clusterProfiler package (version 4.0.5) to convert them to ENTREZID.
We removed all the pathways for which we had included less than 20% of the total amount of genes annotated to the pathway in our feature set and ran the enrichment analysis using the ‘run_enrichment’ method implemented in the R/Bioconductor package MOFA2 (version 1.2.2) with set.statistic parameter ‘rank.sum’ and default parameters otherwise. We ran the enrichment separately for features with only positive or negative weights and jointly across all features.
Pathways with an adjusted P < 0.05 (Benjamini–Hochberg adjustment) have been considered to be significantly enriched.
Cell–cell communication
To analyze the potential axes of cell–cell communication between different cell types, we used the previous knowledge about potential ligand–receptor–target interactions of the NicheNet21 resource collected in the nichenetr package (version 1.1.0) and loaded the provided ligand–receptor network and ligand–target matrix66. Based on the classifications in those networks, we identified ligands, receptors and potential targets among the 13,382 features included in our integrated dataset resulting from the ‘data harmonization and integration step’. We calculated Spearman correlation between all identified ligand–target pairs within this dataset.
For the further analysis of the calculated ligand–target correlations in combination with the corresponding regulatory potential score, we only considered ligand–target pairs:
-
Between the ligands and targets of different cell types (for example, between monocytes and T cells) and different views (for example, between cytokines and the different cell-type clusters)
-
Where we have reliably measured a receptor in the target cell-type cluster to which the ligand might potentially bind as specified by ligand–receptor network provided by the NicheNet21 resource to affect the target gene (in case the target belongs to one of the cell-type cluster views from the scRNA-seq data). We consider a receptor gene to be reliably measured in case it fulfills one of the thresholds below:
-
Percentage of cells expressing the receptor gene in the cell-type cluster > 30 ∩ total amount of cells expressing the receptor gene in cell-type cluster > 600
-
Percentage of cells expressing the receptor gene in the cell-type cluster > 10 ∩ total amount of cells expressing the receptor gene in the cell-type cluster > 1,200
-
Subsequently, we focused on pairs with high correlation and regulatory potential scores where the target gene has a high feature weight on the analyzed MOFA factor.
For the lagged analysis of ligand–receptor target gene interactions, we applied the same analysis with the modification that we calculated Spearman correlation between the identified ligand–target pairs based on mapping lagged ligand expression to the corresponding target gene expression for ACS samples. This resulted in the following mapping across timepoints for each patient that was then used for the calculation of Spearman correlation and further processing as explained above:
Ligand gene | TP1M | TP2M | TP3M |
Target gene | TP2M | TP3M | TP4M |
Predictions
To evaluate the prediction potential of our MOFA factors to distinguish our different patient groups, we calculated ROC curves contrasting the prediction power of the inferred factors to established clinical markers.
For Factor 4 predictions, we only considered factor values from samples measured at TP1M that could be classified to have a ‘good’ or ‘poor’ outcome. We compared the prediction potential of the factor values to the value of the clinical markers (CK, troponin, CRP) for those samples at TP1M. For the benchmarking against the prediction power across the complete time course of the clinical values, we took the maximum and mean values of those markers across all measured timepoints. In both cases, we scaled the clinical values as well as the factor values to be in a range between 0 and 1 and used them as input for the ROC curve calculation giving the probability of a sample being classified as ‘good’ versus ‘poor’ outcome.
Replication in the validation cohort: Groningen dataset
To validate our approach and findings, we used an independent second external dataset including n = 24 patients measured across three different timepoints (TP1G, at the heart catheterization; TP2G, 24 h after admission; TP3G, after 6–8 weeks) and a control group (TP0G, n = 31) contained within the Groningen study and described in detail in the original publication12. Data collected in the Groningen cohort were part of the CardioLines biobank67 study that investigated factors influencing diagnosis and treatment outcomes. It was approved by the UMCG ethics committee (document number METC UMCG 2012/296), and all patients provided informed consent68,69.
Further specifications of the dataset and the processing can be found within the corresponding paper12. The data in the Groningen study12 were measured with two different chemistries: v2 10X chemistry and v3 10X chemistry, which showed technical differences in gene expression profiles between the samples that were prepared with different chemistries. Therefore, a separate processing of both datasets was necessary. In our replication, we focused on samples measured with the v2 10X chemistry as this cohort included a higher number of samples (v2, n = 55; v3, n = 21). The V3 10X chemistry cohort did not include a sufficient number of samples that could be divided into poor or good outcome groups and would have therefore been underpowered. Classification into long-term good- and poor-outcome groups in the Groningen cohort was performed to subdivide the cohort into two outcome sub-cohorts based on long-term development of EF. Based on the ΔEF from echocardiography results (during the hospital stay and follow-up), a classification was made according to positive (good outcome, n = 7) and negative or stable (poor outcome, n = 5) long-term values.
For the replication, in a first step, we evaluated the alignment of the different strategies for cell-type annotations that were applied in the two different studies. Subsequently, we harmonized annotations between both datasets and applied the Munich data preprocessing strategy on the Groningen data. MOFA Factor 1 (IC) could not be evaluated in the Groningen cohort as this dataset does not include the differentiation of the control group into ‘CCS’ and ‘non-CCS’ patients.
Alignment of cell-type annotations
In the Groningen study, cell-type annotation was done using the automated Azimuth method from the R Azimuth library (version 0.4.6; for more details, see paper12). As our data were processed and annotated in a different way, we first compared clusters and annotations resulting from our study to those of the Groningen study. For this, we ran the preprocessing and automated annotation strategy as described in the Groningen study12 on our data and compared the resulting annotations of the single cells with the annotations resulting from our initial clustering and manual annotation strategy (Supplementary Fig. 4). In general, our clustering and the automated azimuth annotation resulted for some cell types in more granular (for example, B cell cluster 10 would be distributed across ‘B naive’, ‘B memory’ and ‘B intermediate’ azimuth annotations) or more aggregated annotations (for example, CD14high monocyte clusters 4, 6 and 7 would all be aggregated as CD14high monocytes), but on a more aggregated level, annotations aligned well except for some T cell clusters (namely, cluster 1 CD8+ T cells, cluster 11 CD4+ T cells and cluster 5 CD4+ T cells).
Rerun of MOFA with harmonized cell-type annotations
As described in the section ‘Alignment of cell-type annotations’, the Munich and Groningen datasets initially were annotated using two different strategies. To be able to apply a model trained on the Munich cohort to the data from Groningen, the same harmonized definition of cell types and features needed be used in both datasets. Thus, we applied the Azimuth automated cell type annotation approach that was used in the Groningen cohort to the data from the Munich cohort. This led to a new annotation with a different level of granularity, which might impact on the downstream MOFA. To assess this effect, we ran the same MOFA as outlined above on the Munich data with the new Azimuth annotations (Supplementary Fig. 5a). We evaluated whether the resulting factors were able to capture the same patterns as found with our original strategy and whether factor and feature weights of the newly inferred factors and the factors presented in the paper aligned well by correlating them (Supplementary Figs. 5b and 6a,b). Overall, the same patterns presented previously were also visible with the alternative annotations (Supplementary Fig. 5b) and the inferred factor and feature weights of the presented factors were highly correlated (|cor| > 0.8; Supplementary Fig. 6a,b).
Processing of the Groningen scRNA-seq data
In the next step, we applied the preprocessing steps as described in ‘CS scRNA-seq data’ for the MOFA analysis also on the Groningen scRNA-seq dataset resulting in normalized pseudo-bulk-aggregated features per annotated azimuth cell type in the Groningen data.
As the expression values of the genes were notably lower in the Groningen dataset, we made some minor adjustments to the requirements with regard to the percentage of cells expressing a gene and the total amount of cells expressing a gene to get a comparable set of features as in our data:
-
Percentage of cells expressing gene > 30 ∩ total amount of cells expressing gene > 1,000
-
Percentage of cells expressing gene > 20 ∩ total amount of cells expressing gene > 2,500
After applying these preprocessing steps on the Groningen data, we had in total 6,353 features across the 13 different dimensions (azimuth cell types 1–13).
Factor 2: time pattern replication
To replicate Factor 2 on the Groningen dataset in a first step, we mapped input features from the Munich dataset to the Groningen dataset and kept only features available in both datasets after the preprocessing (therefore, cytokine, clinical, proteomics and neutrophil features as well as some genes from the scRNA-seq dataset not within both datasets were removed). We used the resulting feature-weight matrix for features from the well-aligned cell types (B cell, CD4TCM, cDC2, CD16Mono, CD14Mono, NK) from the Munich azimuth MOFA estimation (WMU) (Supplementary Fig. 5) and calculated the right inverse to then apply it on the normalized input data (YGR) from the Groningen cohort to infer the corresponding sample factor matrix (ZGR) for the Groningen cohort as shown below. The pattern across timepoints of the resulting sample factor matrix (ZGR) was then compared with the pattern given within the Munich dataset.
GR, Groningen cohort; MU, Munich cohort.
Factor 4: prediction replication
For Factor 4, the main goal of our replication was to evaluate the potential of top-ranking features on Factor 4 to predict the outcome already at an early stage (TP1). As Factor 4 is derived based on the pattern across all four timepoints measured in our data and top-ranking Factor 4 features are characterized not only by the variation at TP1 between ‘good’ and ‘poor’ outcome samples but also by the variation across the different timepoints, we chose to add another step to our analysis to identify those features within the top-ranking features that have high prediction potential only looking at their TP1 values. For this, we chose to select the intersection of the top 280 features of IAR (corresponding to roughly 20% of the total amount of features) between the MOFA models estimated on the Munich data with the manual and the harmonized automated azimuth annotations. Subsequently, we trained a lasso model (logistic regression) with 8-fold cross validation using the cv.glmnet function of the R package glmnet (version 4.1.6; alpha=1; family = ‘binomial’; nfolds = 8; other, default parameters) on the Munich data taking as input only the value of those features at TP1M and predicting the outcome of the samples. We selected the best model given by ‘lambda.min’, the value of lambda that minimizes the cross-validation error.
Then, we applied this trained model on the same set of features with their values at TP1G on the Groningen dataset considering this dataset our holdout test dataset and evaluated prediction performance for those samples calculating AUC values.
In addition to the lasso-selected top-ranking feature set of Factor 4, we also evaluated the prediction potential of features that we highlighted in the paper, based on their potential biological mechanisms, namely NK cell features CD74, TXNIP and GZMB. Again, we trained a logistic regression model (glm function; family = binomial (link = ‘logit’)) for these features on the Munich data. Subsequently, we applied this model to the Groningen cohort for the evaluation of the performance on the validation set.
In vitro treatment of healthy PBMCs with heparin, aspirin and prasugrel with subsequent scRNA-seq
Human PBMCs were isolated from healthy donors as described above and freshly processed for scRNA-seq. A total of 2 × 105 PBMCs were seeded in a 96-well plate and incubated with heparin (1.5 IU ml−1, Ratiopharm), prasugrel (0.012 mg ml−1, Substipharm SAS) and acetylsalicylic acid (0.1 mg ml−1). After 3 h of incubation at 37 °C and 5% CO2, the cells were washed and subsequently treated with Fc-Block (Human BD Fc Block, BD Biosciences, catalog number 564200) for 10 min at 4 °C. Afterward, the respective hashtag master mix (final antibody concentration, 1:100) was added, and the cells were incubated for 30 min at 4 °C. Subsequently, 5 ml of buffer (0.5% BSA (Albumin Fraktion V, 8076.4, Carl Roth) plus DPBS (4190-094, Thermo Fisher)) was added and the mixture was centrifuged at 250 × g for 10 min at 4 °C. This washing step was repeated twice. After the last centrifugation step, the pellet was resuspended in 50 µl buffer (0.5% BSA (Albumin Fraktion V, Carl Roth, catalog number 5642008076.4)). Cell counts were adjusted to 1,000 cells µl−1 using a Neubauer counting chamber and then pooled. A total of 60 µl of the single-cell suspension was used for library preparation (input, 60,000 cells).
Bioinformatic analysis of the in vitro scRNA-seq dataset
For the analysis of the effect of medication on the identified MOFA factors and the underlying expression changes of individual genes, we preprocessed the in vitro (IV) scRNA-seq data in the same way as described within the sections for the preprocessing of the CS dataset: ‘SC data preparation: cellranger’, ‘Demultiplexing and doublet identification’ and ‘Cell quality control and filtering’.
Subsequently, we took the log-normalized data, identified highly variable genes (scanpy.pp.highly_variable_genes; default parameters), regressed out the effects of total counts and the percentage of mitochondrial gene counts (scanpy.pp.regress_out; default parameters) and scaled the gene expression values (sc.pp.scale; max_value = 10). We used these data to do a principal component analysis of the variables (sc.tl.pca; default parameters) and used the embedding of the first 50 principal components as input for the computation of a neighborhood graph (scanpy.pp.neighbors; n_neighbors=10, n_pcs=50) and the subsequent clustering of the cells using the Leiden algorithm (scanpy.tl.leiden; Resolution=0.25; default parameters). The resulting clusters were visualized in a UMAP plot (Supplementary Fig. 9a). For cell-type annotation, we ran the automated Azimuth strategy as described for the replication on the Groningen dataset and compared them with the identified clusters on the same UMAP plot (Supplementary Fig. 9a).
In the next step, we applied the preprocessing steps as described within ‘CS scRNA-seq data’ for the MOFA also on the IV scRNA-seq dataset resulting in normalized pseudo-bulk-aggregated features per annotated azimuth cell type in the IV scRNA-seq data.
As the number of samples in the IV scRNA-seq dataset was lower compared with that in the CS dataset, we made some adjustments to the requirements with regard to the total amount of cells expressing a gene to get a comparable set of features to those in the CS data:
-
Percentage of cells expressing gene > 50 ∩ total amount of cells expressing gene > 60
-
Percentage of cells expressing gene > 40 ∩ total amount of cells expressing gene > 150
After applying these preprocessing steps on the IV scRNA-seq data, we had in total 13,676 features across the 13 different views (azimuth cell types 1–13, YIV).
To evaluate the effect of the medication on Factor 2 values, we computed Factor 2 on the IV dataset by applying the same strategy as in the Groningen replication: first, we mapped input features from the Munich CS dataset to the Munich IV dataset and kept only features available in both datasets after applying the preprocessing as indicated. We used the feature-weight matrix from the Munich CS azimuth MOFA estimation (WMU) and calculated the right inverse to then apply it on the normalized input data (YIV) from the IV scRNA-seq data to infer the corresponding sample factor matrix (ZIV) for the IV dataset. We then evaluated the difference of Factor 2 values between medication-treated and untreated samples (Supplementary Fig. 9b).
In addition, we also investigated how effect sizes of ACS at TP2M compared with those of CCS of single genes that we outlined in the main figures by violin plots compared with the effect sizes introduced by the medication, comparing treated samples with untreated samples. For that, we took the pseudo-bulk-aggregated and normalized gene expression based on the azimuth annotation of the Munich IV scRNA-seq dataset (\({Y}_{i}^{\;\mathrm{IV}}\)) and the Munich CS dataset (\({Y}_{i}^{\;\mathrm{CS}}\)) and calculated a linear model estimating the effect of the medication treatment (βT) compared with the untreated samples and the effect of ACS at TP2M (βTP2M) compared with the CCS samples, correspondingly (Supplementary Fig. 9c).
For the comparison, we evaluated the effect sizes of the pseudo-bulk-aggregated gene expression values for cells annotated as CD4.TCM or CD14.Mono cells according to the azimuth annotation as these aligned best with the clusters (cluster 0 CD4+ T cells; cluster 4, 6 and 7 CD14high monocytes) in which we observed the effects of ACS TP2M outlined in the main figures (Fig. 3c,d).
Echocardiographic assessment of left ventricular function in the Munich cohort
Echocardiographic assessment of left ventricular EF as a proxy for systolic function was performed according to current guidelines70. In brief, B-mode echocardiography was performed by cardiac intensive care unit (C-ICU) fellows and, following discharge from the ICU, by cardiology residents. Left ventricular EF was then measured using the biplane summation-of-disks method in apical four-chamber and apical two-chamber views by an experienced cardiology fellow who was unaware of patient outcomes at the time of measurement.
IV assays
IV monocyte survival, apoptotic Jurkat cell generation and monocyte efferocytosis assay
PBMCs from healthy donors who gave informed consent were used (with the approval of the Ethics Committee of LMU Munich (number 19-274)) using BD Vacutainer CPT tubes and subsequently stored at −80 °C. Monocytes were isolated (CD14 MicroBeads, Miltenyi), kept in monocyte RPMI 1640 medium (Sigma-Aldrich, catalog number R8758) supplemented with 10% FBS (Biosell, catalog number S0613) and 1% penicillin–streptomycin (Sigma-Aldrich, catalog number P4333), and subsequently labeled with CellTracker green (CMDFA, catalog number C7025), and cells were seeded in a 24-well plate at a concentration of 100,000 cells per well. Selective gp130 (IL6ST) inhibitor SC144 10 µM (MedChemExpress, catalog number HY-15614) in 1% DMSO or DMSO 1% (Sigma-Aldrich, catalog number D2438-10ML) was supplemented to the different conditions for the inhibitor experiments. For patient plasma assays, diluted plasma from randomly selected patients from the Munich cohort with CCS (TP0M) or sterile ACS (TP1M–TP4M) (1:1 dilution with RPMI 1640 (Sigma-Aldrich, catalog number R8758)) was added to the respective wells. Images were taken at timepoints 0 h and 24 h.
Efferocytosis assay was performed similarly to the murine macrophage efferocytosis assay previously described71: in short, cultured Jurkat cells (Merck, catalog number 88042803) were centrifuged and resuspended at a concentration of 2.5 × 105 ml−1. Apoptosis was induced using UV light radiation for 15 min. Next, Jurkat cells were placed in the incubator for 3.5 h. A total of 100,000 apoptotic Jurkat cells (labeled with far-red cell Proliferation Dye eFluor 670 (eBioscience, catalog number 65-0840-85)) were added to the monocytes 24 h after addition of the selective gp130 (IL6ST) inhibitor SC144, 10 µM (MedChemExpress, catalog number HY-15614) in 1% DMSO or only DMSO 1% (Sigma-Aldrich, catalog number D2438), and 24 h after adding the respective diluted patient plasma. Cells were subsequently co-incubated for 8 h and washed before image acquisition. Cells were incubated at 37 °C and 5% CO2. The resulting images were analyzed in FIJI (ImageJ) using the automated cell counting function.
IV monocyte chemotaxis
Human monocytes were isolated as described above. Following counting, 2 × 105 monocytes were seeded into a 96-well plate and treated with either DMSO 1% (Sigma, catalog number D2438) or SC144 (10 µM, Selleckchem, catalog number S7124) in monocyte medium for 2 h at 37 °C and 5% CO2. For chemotaxis, after the incubation period, 1 × 105 monocytes were stained using CellTracker Red CMTPX (1:1,000, Invitrogen, catalog number C34552) for 10 min. Subsequently, monocytes were allowed to attach to the upper part of a 5 µm transwell insert (Sarstedt), and the transwell insert was transferred to an ultra-low-attachment 24-well plate (Corning) containing monocyte medium (mentioned above) mixed with CCL2 (100 µg ml−1, Bio-Techne, catalog number 279-MC/CF). After incubation for 2 h at 37 °C and 5% CO2, cells were harvested from the lower chamber. To allow further detachment of cells, 5 mM EDTA was added. Subsequently, the number of transmigrated cells was measured with flow cytometry and standardized to counting beads (CountBright, Invitrogen).
IV monocyte ROS production
Following counting, 2 × 105 monocytes were seeded into a 96-well plate and treated with either DMSO 1% (Sigma, catalog number D2438) or SC144 (10 µM, Selleckchem, catalog number S7124) in monocyte medium for 2 h, or were treated with randomly selected CCS (TP0M) or sterile ACS plasma at different timepoints for 12 h at 37 °C and 5% CO2. Afterward, monocytes were loaded with 2′,7′-dichlorodihydrofluorescein diacetate (2′,7′ DCFDA; 5 µM, Sigma) and incubated for 15 min at 37 °C and 5% CO2. Subsequently, cells were exposed to PMA (200 nM, Sigma) for 1 h at 37 °C and 5% CO2, followed by immediate flow cytometric analysis. SYTOX Red (1:1,000, Invitrogen, catalog number S34859) was added for live or dead staining.
IV monocyte phenotyping
Human monocytes were isolated from healthy donors as described above. A total of 1 × 105 monocytes were seeded in a 96-well plate and co-incubated with plasma from randomly selected patients with CCS (TP0M) or sterile ACS (at the different timepoints: TP1M–TP4M) either in the presence of anti-hIL-6 (1 µg ml−1, Invivogen, catalog number mabg-hil6-3) or isotype control (1 µg ml−1, Invivogen, catalog number mabg1-ctrlm), or in the absence of any additional treatment. After a 12 h incubation at 37 °C and 5% CO2, the cells underwent a washing step and were then incubated with Fc-Block (1:100, BD Biosciences, catalog number 564220) for 10 min, followed by incubation with the respective primary antibodies (1:100; Supplementary Table 15) for 20 min on ice in the dark. Following another wash step, flow cytometric analysis was conducted. Before FACS, live and dead staining was added.
IV T cell phenotyping
After isolation (as described above), PBMCs were stained with carboxyfluorescein succinimidyl ester proliferation dye according to the manufacturer’s instruction (Invitrogen, catalog number C34554). Upon completion of the staining, cells were resuspended in RPMI 1640 medium supplemented with 10% FBS, l-glutamine, nonessential amino acids, sodium pyruvate and penicillin–streptomycin, and plated in 96-well flat-bottom plates.
Plasma derived from randomly selected patients with CCS (TP0M) or sterile ACS (at the different timepoints: TP1M–TP4M) was then added to each well at a plasma ratio of 1:3. PBMCs were incubated with the respective plasma for 96 h. T cell activation and phenotypical changes were then analyzed by flow cytometry, and the average of at least five PBMC donors was calculated per incubated plasma.
Flow cytometry was performed as recently described72,73. In brief, after 96 h of incubation of isolated PBMCs with plasma, cells were transferred into a 96-well round-bottom plate and centrifuged for 5 min at 400 × g. Cells were then washed twice with ice-cold PBS and incubated with human TrueStain FcX and fixable viability dye (eFluor 780, eBioscience) for 15 min at room temperature. Next, cells were stained with the respective antibodies (Supplementary Table 15) for 30 min at 4 °C. After 30 min, cells were again washed with ice-cold PBS fixed with 1% paraformaldehyde solution. Flow cytometric data were analyzed using FlowJo V10.9.0 software.
Monocyte cytokine secretion upon inhibition of IL-6 signaling
Human monocytes were isolated according to the monocyte isolation protocol described above. Following counting, 1 × 105 monocytes were seeded into a 96-well plate, treated with either DMSO (1%, Sigma, catalog number D2438) or SC144 (10 µM, Selleckchem, catalog number S7124) and afterward activated with PMA (200 nM, Sigma) and subsequently incubated in monocyte medium (described above) for 18 h at 37 °C and 5% CO2.
After the incubation period, the cells were centrifuged at 350 × g for 10 min and the cell culture supernatant was immediately used for cytokine analysis. Cytokine levels of cultured supernatants were determined with LEGENDplex Human Inflammation Panel 1 (Biolegend, catalog number 740809) according to the manufacturer’s protocol. In short, supernatants were incubated for 2 h at room temperature with capture beads on a plate shaker. After washing, the detection antibodies were added and incubated for another 1 h at room temperature. Once the beads were stained, the samples were measured using a CytoFLEX flow cytometer (Beckman Coulter). Based on the standard curve, the cytokine concentration was calculated using BioLegend’s LEGENDplex data analysis software (analyzed with the newest version in September 2023). For every sample, a duplicate was measured simultaneously, and the mean value was calculated and used for statistical analysis.
Flow cytometry of all analyses in Methods were carried out with BD LSRFortessa.
Statistical analysis
The statistical analysis and the graphical illustration were performed with GraphPad Prism (versions 9.2.0 and 10.0.3). The data were tested for normal distribution using the Shapiro–Wilk normality test. Parametric tests were used only when all involved groups showed a parametric distribution; otherwise, a nonparametric test was used. Parametric-distributed data with several subgroups were analyzed using ordinary one-way ANOVA, with correction for multiple comparisons (for example, the single timepoints) by Dunnett’s test per feature individually. Nonparametric-distributed data with several subgroups were analyzed using the Kruskal–Wallis test, with correction for multiple comparisons (for example, the single timepoints) by Dunn’s test per feature individually. Comparisons between two groups were performed using unpaired t-test (two sided) for parametric data and Mann–Whitney U test (two sided) for nonparametric data. For paired nonparametric analyses, the Wilcoxon rank sum test (two sided) was used; for paired parametric analyses, a paired t-test (two sided) was used. In the good-versus-poor outcome comparison across timepoints, a mixed-effects analysis with correction for multiple comparisons by Šidák test was used. If only three groups were compared, the Tukey test was used to correct for multiple comparison. In case only the mixed-effects analysis, ordinary one-way ANOVA or Kruskal–Wallis test, but not the multiple comparison, was significant, graphs are marked with a vertical bar on top. Graphs in which only the post hoc test was significant were marked in the figure captions.
Figure alignment
Figures were aligned by Adobe Illustrator (version 25.4.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All mapped data (bulk proteomics, cytokine measurements, biomarkers of inflammation and myocardial damage, ejection fraction, bulk and scRNA-seq count matrices) from the Munich cohort are available via Zenodo upon approval for immunological research purposes (https://doi.org/10.5281/zenodo.10815146 (ref. 74)). Processed pathway annotations and auxiliary data are also provided via Zenodo. Raw sequencing data of the Groningen cohort are available from the European Genome–Phenome Archive (https://ega-archive.org/datasets/EGAD00001010064), and processed data are available from https://eqtlgen.org/sc/datasets/blokland2024-dataset.html. Ligand receptor data and regulatory potential scores from the NicheNet model21 were downloaded from https://doi.org/10.5281/zenodo.3260758 (ligand receptor network and regulatory potential scores)66. Reference data for the azimuth mapping were a previously annotated and published multimodal CITE-seq (combined scRNA-seq and protein expression) reference dataset of 162,000 PBMCs75.
Code availability
All code is available via GitHub at https://github.com/heiniglab/stemi_mofa.
References
Sanchis-Gomar, F., Perez-Quilis, C., Leischik, R. & Lucia, A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann. Trans. Med. 4, 256 (2016).
Dalen, J. E., Alpert, J. S., Goldberg, R. J. & Weinstein, R. S. The epidemic of the 20th century: coronary heart disease. Am. J. Med. 127, 807–812 (2014).
Weil, B. R. & Neelamegham, S. Selectins and immune cells in acute myocardial infarction and post-infarction ventricular remodeling: pathophysiology and novel treatments. Front. Immunol. 10, 300 (2019).
Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91–112 (2016).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83 (2017).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Rood, J. E., Maartens, A., Hupalowska, A., Teichmann, S. A. & Regev, A. Impact of the human cell atlas on medicine. Nat. Med. 28, 2486–2496 (2022).
Perez, R. K. et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376, eabf1970 (2022).
Wu, T. D. et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278 (2020).
Argelaguet, R. et al. Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 14, e8124 (2018).
van Blokland I. V. et al. Single-cell dissection of the immune response after acute myocardial infarction. Preprint at medRxiv https://doi.org/10.1101/2023.05.02.23289370 (2023).
Jerby-Arnon, L. & Regev, A. DIALOGUE maps multicellular programs in tissue from single-cell or spatial transcriptomics data. Nat. Biotechnol. 40, 1467–1477 (2022).
Janjic, A. et al. Prime-seq, efficient and powerful bulk RNA sequencing. Genome Biol. 23, 88 (2022).
De Silva, D. et al. Robust T cell activation requires an eIF3-driven burst in T cell receptor translation. eLife 10, e74272 (2021).
Galazka, G. et al. HINT1 peptide/Hsp70 complex induces NK-cell-dependent immunoregulation in a model of autoimmune demyelination. Eur. J. Immunol. 44, 3026–3044 (2014).
Li, G., Liang, X. & Lotze, M. T. HMGB1: the central cytokine for all lymphoid cells. Front. Immunol. 4, 68 (2013).
Szabo, P. A. et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 10, 4706 (2019).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Kanehisa, M. The KEGG database. Novartis Found. Symp. 247, 91–101 (2002).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Schröder, B. The multifaceted roles of the invariant chain CD74—more than just a chaperone. Biochim. Biophys. Acta 1863, 1269–1281 (2016).
Bianchi, M., Crinelli, R., Arbore, V. & Magnani, M. Induction of ubiquitin C (UBC) gene transcription is mediated by HSF1: role of proteotoxic and oxidative stress. FEBS Open Bio. 8, 1471–1485 (2018).
Jiang, F., Gao, Y., Dong, C. & Xiong, S. ODC1 inhibits the inflammatory response and ROS-induced apoptosis in macrophages. Biochem. Biophys. Res. Commun. 504, 734–741 (2018).
Yurdagul, A. Jr. et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 31, 518–533.e10 (2020).
Yang, S., Liu, F., Wang, Q. J., Rosenberg, S. A. & Morgan, R. A. The shedding of CD62L (L-selectin) regulates the acquisition of lytic activity in human tumor reactive T lymphocytes. PLoS ONE 6, e22560 (2011).
Chao, C. C., Jensen, R. & Dailey, M. O. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 159, 1686–1694 (1997).
Patsoukis, N., Wang, Q., Strauss, L. & Boussiotis, V. A. Revisiting the PD-1 pathway. Sci. Adv. 6, eabd2712 (2020).
Sabins, N. C. et al. TIM-3 engagement promotes effector memory T cell differentiation of human antigen-specific CD8 T cells by activating mTORC1. J. Immunol. 199, 4091–4102 (2017).
Swirski, F. K. & Nahrendorf, M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744 (2018).
Fox, K. A. et al. Should patients with acute coronary disease be stratified for management according to their risk? Derivation, external validation and outcomes using the updated GRACE risk score. BMJ Open 4, e004425 (2014).
Kim, D. O. et al. TXNIP regulates natural killer cell-mediated innate immunity by inhibiting IFN-γ production during bacterial infection. Int. J. Mol. Sci. 21, 9499 (2020).
Voskoboinik, I., Whisstock, J. C. & Trapani, J. A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 (2015).
Tang, X., Metzger, D., Leeman, S. & Amar, S. LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced LPS-induced cytokine: evidence for LITAF-dependent LPS signaling pathways. Proc. Natl Acad. Sci. USA 103, 13777–13782 (2006).
Lowin-Kropf, B., Kunz, B., Schneider, P. & Held, W. A role for the src family kinase Fyn in NK cell activation and the formation of the repertoire of Ly49 receptors. Eur. J. Immunol. 32, 773–782 (2002).
Perišić Nanut, M., Sabotič, J., Švajger, U., Jewett, A. & Kos, J. Cystatin F affects natural killer cell cytotoxicity. Front. Immunol. 8, 1459 (2017).
Todros-Dawda, I., Kveberg, L., Vaage, J. T. & Inngjerdingen, M. The tetraspanin CD53 modulates responses from activating NK cell receptors, promoting LFA-1 activation and dampening NK cell effector functions. PLoS ONE 9, e97844 (2014).
Altara, R., Mallat, Z., Booz, G. W. & Zouein, F. A. The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J. Immunol. Res. 2016, 4396368 (2016).
Wang, Z. et al. CCL24/CCR3 axis plays a central role in angiotensin II-induced heart failure by stimulating M2 macrophage polarization and fibroblast activation. Cell. Biol. Toxicol. 39, 1413–1431 (2022).
Bohdziewicz, A. et al. Future treatment options in systemic sclerosis—potential targets and ongoing clinical trials. J. Clin. Med. 11, 1310 (2022).
Aslam, M. S., Aslam, M. S., Aslam, K. S., Iqbal, A. & Yuan, L. Therapeutical significance of Serpina3n subsequent cerebral ischemia via cytotoxic granzyme B inactivation. Biomed. Res. Int. 2022, 1557010 (2022).
Montoya, M. C. et al. Role of ICAM-3 in the initial interaction of T lymphocytes and APCs. Nat. Immunol. 3, 159–168 (2002).
Szymczak-Workman A. L., Jones L. L., Guy C. S., Vignali D. A. A. & Kitainda V. in Encyclopedia of Biological Chemistry III 3rd edn (ed. Jez, J.) 294–301 (Elsevier, 2021).
Samara, P., Ioannou, K. & Tsitsilonis, O. E. Prothymosin alpha and immune responses: are we close to potential clinical applications? Vitam. Horm. 102, 179–207 (2016).
Travelli, C., Colombo, G., Mola, S., Genazzani, A. A. & Porta, C. NAMPT: a pleiotropic modulator of monocytes and macrophages. Pharmacol. Res. 135, 25–36 (2018).
Halvorsen, B. et al. Increased expression of NAMPT in PBMC from patients with acute coronary syndrome and in inflammatory M1 macrophages. Atherosclerosis 243, 204–210 (2015).
Saigusa, R. et al. Sex differences in coronary artery disease and diabetes revealed by scRNA-seq and CITE-seq of human CD4+ T cells. Int. J. Mol. Sci. 23, 9875 (2022).
McCaffrey, T. A. et al. RNA sequencing of blood in coronary artery disease: involvement of regulatory T cell imbalance. BMC Med. Genomics 14, 216 (2021).
Leistner, D. M. et al. Differential immunological signature at the culprit site distinguishes acute coronary syndrome with intact from acute coronary syndrome with ruptured fibrous cap: results from the prospective translational OPTICO-ACS study. Eur. Heart J. 41, 3549–3560 (2020).
Ruggio, A. et al. Correlation between CD4+CD28null T lymphocytes, regulatory T cells and plaque rupture: an optical coherence tomography study in acute coronary syndromes. Int. J. Cardiol. 276, 289–292 (2019).
Santos-Zas, I. et al. Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 12, 1483 (2021).
Yan, X. et al. Lung natural killer cells play a major counter-regulatory role in pulmonary vascular hyperpermeability after myocardial infarction. Circ. Res. 114, 637–649 (2014).
Selathurai, A. et al. Natural killer (NK) cells augment atherosclerosis by cytotoxic-dependent mechanisms. Cardiovasc. Res. 102, 128–137 (2014).
Lu, J. et al. Multi-omics reveals clinically relevant proliferative drive associated with mTOR-MYC-OXPHOS activity in chronic lymphocytic leukemia. Nat. Cancer 2, 853–864 (2021).
Pekayvaz, K. et al. Protective immune trajectories in early viral containment of non-pneumonic SARS-CoV-2 infection. Nat. Commun. 13, 1018 (2022).
Hughes, C. S. et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014).
Bruderer, R. et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 (2015).
Andrews S. FastQC: a quality control tool for high throughput sequence data (Babraham Institute, 2010); http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Patzelt, J. & Langer, H. F. Platelets in angiogenesis. Curr. Vasc. Pharmacol. 10, 570–577 (2012).
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. & Hellmann, I. zUMIs—a fast and flexible pipeline to process RNA sequencing data with UMIs. GigaScience 7, giy059 (2018).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Fresh Frozen Human Peripheral Blood Mononuclear Cells for Single Cell RNA Sequencing Document Number CG00039 Rev E (10x Genomics, 2021); https://assets.ctfassets.net/an68im79xiti/71r5PbRPB1LeqRkuPltBzr/64cfaa099d0a7fd41f79a4aecd643926/CG00039_Demonstrated_Protocol_FreshFrozenHumanPBMCs_RevE.pdf
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Browaeys, R., Saelens, W. & Saeys, Y. Development, evaluation and application of NicheNet: datasets. Zenodo https://doi.org/10.5281/zenodo.3260758 (2019).
van Blokland, I. V., Groot, H. E., Hendriks, T., Assa, S. & van der Harst, P. Sex differences in leukocyte profile in ST-elevation myocardial infarction patients. Sci. Rep. 10, 6851 (2020).
Tigchelaar, E. F. et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5, e006772 (2015).
Oelen, R. et al. Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure. Nat. Commun. 13, 3267 (2022).
Mitchell, C. et al. Guidelines for performing a comprehensive transthoracic echocardiographic examination in adults: recommendations from the American Society of Echocardiography. J. Am. Soc. Echocardiogr. 32, 1–64 (2019).
Pekayvaz, K. et al. Mural cell-derived chemokines provide a protective niche to safeguard vascular macrophages and limit chronic inflammation. Immunity 56, 2325–2341.e15 (2023).
Gottschlich, A. et al. Single-cell transcriptomic atlas-guided development of CAR-T cells for the treatment of acute myeloid leukemia. Nat. Biotechnol. 41, 1618–1632 (2023).
Wagenbauer, K. F. et al. Programmable multispecific DNA-origami-based T-cell engagers. Nat. Nanotechnol. 18, 1319–1326 (2023).
Pekayvaz, K. et al. Multiomic analyses uncover immunological signatures in acute and chronic coronary syndromes. Zenodo https://doi.org/10.5281/zenodo.10815146 (2024).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Acknowledgements
We thank the patients and participants for participating in the study. This study was supported by the DZHK partner site project (K. Stark), DZHK Säule B Antrag DZHK B 21-014 SE (K.P., L.N., P.M. and N.H.) and DZHK project 81Z0600106 (M.H.). Furthermore, the project was supported by Deutsche Herzstiftung e.V., Frankfurt a.M. (K.P. and L.N.), Deutsche Forschungsgemeinschaft (DFG) SFB 914 (S.M. (B02 and Z01) and K. Stark (B02)), the DFG SFB 1123 (S.M. and L.N. (B06), K. Stark (A07), and M.J and R.Z. (Z02)), the DFG SFB1321 (S.M. (P10), the DFG FOR 2033 (S.M.), the DFG SFB 359 (K. Stark (A03)), the DFG SFB1243 (W.E. (A14)), the DFG EN 1093/2-1 (W.E. and A.J.), individual funding from the DFG (PE 2704/3-1) to T.P., the DFG NI 2219/2-1 to L.N. and the DFG GO 3823/1-1 to A.G.; grant numbers 510821390, KO5055-2-1 and KO5055/3-1 to S.K., LMUexcellent (K.P.), the Else-Kröner-Fresenius Stiftung (K.P.), the Corona Foundation (L.N.), the DFG Clinician Scientist Programme PRIME (413635475, K.P. and R.K.), the ERC-Starting grant ‘T-MEMORE’ (ERC grant 947611 (K. Stark)) and the ERC Advanced grant ‘IMMUNOTHROMBOSIS’ (ERC-2018-ADG (S.M.)) and the German Centre for Cardiovascular Research (DZHK) (Clinician Scientist Programme (L.N.), MHA 1.4VD (S.M.)), Förderprogramm für Forschung und Lehre der Medizinischen Fakultät der LMU (grant number 1138 to A.G.), the Bavarian Cancer Research Center (BZKF) (A.G. and TANGO to S.K.), the international doctoral program ‘i-Target: immunotargeting of cancer’ (funded by the Elite Network of Bavaria; to S.K.), the Melanoma Research Alliance (grant number 409510 to S.K.), Marie Sklodowska-Curie Training Network for Optimizing Adoptive T Cell Therapy of Cancer (funded by the Horizon 2020 programme of the European Union; grant 955575 to S.K.), Else Kroner-Fresenius-Stiftung (to A.G. and IOLIN to S.K.), German Cancer Aid (AvantCAR.de to S.K.), the Wilhelm-Sander-Stiftung (to S.K.), Ernst Jung Stiftung (to S.K.), Institutional Strategy LMUexcellent of LMU Munich (within the framework of the German Excellence Initiative to S.K.), the Go-Bio-Initiative (to S.K.), the m4-Award of the Bavarian Ministry for Economical Affairs (to S.K.), Bundesministerium fur Bildung und Forschung (to S.K.), European Research Council (Starting Grant 756017 and PoC Grant 101100460 to S.K.), by the SFB-TRR 338/1 2021–452881907 (to S.K.), Fritz-Bender Foundation (to S.K.), Deutsche José Carreras Leukämie Stiftung (to S.K.), Hector Foundation (to S.K.), the Ernst und Berta Grimmke Stiftung (L.N.), the GTH Junior research grant (L.N.), the Bavarian Research Foundation (BAYCELLATOR to S.K.) and the Bruno and Helene Jöster Foundation (360° CAR to S.K.). G.V.H. was supported by a grant from the Förderprogramm fur Forschung und Lehre (FöFoLe) of LMU Munich. C.L. is supported by the Helmholtz Association under the joint research school ‘Munich School for Data Science – MUDS’. N.H. is supported by the DFG SFB-1470-B03, the Chan Zuckerberg Foundation and an ERC Advanced Grant under the European Union Horizon 2020 Research and Innovation Program (AdG788970). M.H. is supported by the Chan Zuckerberg Foundation.
Author information
Authors and Affiliations
Contributions
Initiation and experimental design: K.P., L.N. and K. Stark. Project administration and supervision: K.P., M.H., L.N. and K. Stark. Conceptualization: K.P., C.L., V.K., M.H., L.N. and K. Stark. Writing of the original draft: K.P. Patient identification, patient information and informed consent acquisition, and sample acquisition: K.P., T.P., L.N. and K. Stark. Methodology, investigation and formal analysis: K.P., V.K., C.L., C.G., A.G., G.V.H., A.M.-N., N.B., S.S., S.A., K.Y., S.K., L.N., M.J., S.B., V.K., L.E., R.K., I.V.v.B., R.O., H.E.G., J.W.B., A.J., V.P., R.E., K. Stark, O.P. and M.H. Writing and editing: all authors. Visualization: C.L. and V.K. Funding acquisition: K.P., N.H., P.M., K. Saar, N.H., P.v.d.H., L.H.F., M.G.P.v.d.W., W.E., R.Z., A.G., S.K., S.M., M.H., L.N. and K. Stark.
Corresponding authors
Ethics declarations
Competing interests
S.K. has received honoraria from TCR2, Novartis, BMS, Galapagos, Miltenyi and GSK. S.K. received license fees from TCR2 and Carina Biotech. A.G. received research support from Tabby Therapeutics for work unrelated to the paper. S.K. received research support from TCR2, Plectonic, Catalym and Arcus Bioscience for work unrelated to the paper. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Matthew Feinstein, Manuel Mayr and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Michael Basson, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Changes in clinical values and cell-type abundance across time.
(a) Clinical blood tests. Individual timepoints for sterile ACS (TP1-4M) compared to CCS (TP0M). CRP; Neutrophils. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. Mean values with +/- SEM are shown. (b) Analysis of centered log ratio (CLR) transformed cell type abundance of phenotypically defined immune cells to CD45+ leukocytes in PBMCs based on flow cytometry. Individual timepoints of sterile ACS compared to CCS. Parametric distributed data were analyzed using the ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. Mean values with +/- SEM are shown. In case only the ordinary one-way ANOVA or Kruskal–Wallis test was significant, graphs are marked with a vertical bar on top. (c) Analysis of centered log ratio (CLR) transformed cell type abundance based on scRNA-seq dataset. Individual timepoints of sterile ACS compared to patients with CCS. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. For the following comparisons, only the post hoc was significant: CD4+ T cells – cluster 5. *p≤0.05). Data are shown as a Box-Whiskers plot (box: median, 25th to 75th percentile; whiskers: minimum to maximum). (a)-(c) Exact p-values (Supplementary Table 13) and n-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 2 Gene expression changes across time and disease states in IAI.
(a) Integrative ACS Ischemia (Factor 2). Normalized expression values of top 1% features for sterile ACS and CCS patients in longitudinal comparison visualized within heatmap and weight of the features visualized as barplot (+/- indicates the direction of the feature factor weight). N-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 3 In vitro ACS plasma induced changes in monocyte phenotype and function.
(a) Monocytes isolated from healthy donors incubated with patient plasma, and phenotyped by flow cytometry. Individual timepoints of sterile ACS compared to CCS. Parametric distributed data were analyzed using the ordinary one-way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal–Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05. In case only the ordinary one-way ANOVA or Kruskal–Wallis test was significant, graphs are marked with a vertical bar on top. Mean values with +/- SEM are shown. (b) Monocyte efferocytosis, ROS production and survival after plasma incubation. Individual timepoints of sterile ACS compared to CCS. Non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. Mean values with +/- SEM are shown. (a) – (b) Exact p-values (Supplementary Table 13) and n-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 4 Analysis of medication mediated effects on IAI.
(a) Longitudinal comparison of the factor value of Integrative ACS Ischemia between patients with sterile ACS with and without pre-medication. ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. The dataset was analyzed using the Mixed-effects analysis with correction for multiple comparisons by Šidák test. *p≤0.05. Mean values with +/- SEM are shown. (b) Integrative ACS Ischemia (Factor 2). Comparison of the factor values of each timepoint of sterile ACS (TP1-4M) and non-CCS (TP0M) with CCS (TP0M) patients. Only patients without pre-medication (upper part) of: ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. Only patients with pre-medication (lower part) of: ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. Mean values with +/- SEM are shown. (c) Comparison of the factor value (Integrative ACS Ischemia, Factor 2) in patients without ACS (pooled non-CCS and CCS) between with and without respective pre-medication: ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. The parametric dataset was analyzed using an unpaired t-test (two-sided). *p≤0.05. Mean values with +/- SEM are shown. (d) Integrative ACS Ischemia (Factor 2). Comparison of the factor values of TP1-3M with TP4M of patients with sterile ACS. Only patients without pre-medication (upper part) of: ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. Only patients with pre-medication (lower part) of: ASA, ß-Blocker, ACE/AT1-inhibitor, Statin. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. Mean values with +/- SEM are shown. (a) – (d) Exact p-values (Supplementary Table 13) and n-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 5 The effects of IL6ST inhibition on monocyte-driven cytokine secretion.
(a) Monocyte cytokine secretion upon inhibition of IL6 signaling analyzed by flow cytometry. Comparison between control and treatment with IL6ST-inhibitor group (paired dataset, n=8). Parametric data were analyzed using the paired t-test (two-sided), non-parametric data were tested using the Wilcoxon test (two-sided). *p≤0.05, **p≤0.01. Illustration of paired data by connecting the data points. Exact p-values were summarized in Supplementary Table 13.
Extended Data Fig. 6 Cytokine driven or further effects of ACS plasma on immune cells.
(a) Monocyte phenotyping after incubation with sterile ACS (TP1-3M) or CCS (TP0M) plasma upon IL6 inhibition by flow cytometry. All MFIs were normalized to the respective CCS plasma average of the marker. Comparison between isotype and IL6 inhibition (paired dataset). Parametric data were analyzed using the multiple paired t-test (two-sided). *p≤0.05, **p≤0.01, ***p≤0.001. Illustration of paired data by connecting the data points. (b) T cell phenotyping after incubation with sterile ACS (TP1-4M) or CCS (TP0M) plasma by flow cytometry. Individual timepoints of sterile ACS plasma (randomized n=8) compared to CCS, mean value of at least 5 PBMC donors incubated with each patient plasma. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. In case only the ordinary one-way ANOVA or Kruskal–Wallis test was significant, graphs are marked with a vertical bar on top. For the following comparisons, only the post hoc was significant: T effector memory: CD4+ CCR7- CD45RO+. *p≤0.05, **p≤0.01. Mean values with +/- SEM are shown. (a) – (b) Exact p-values (Supplementary Table 13) and n-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 7 Lagged plasma–cell or cell–cell-mediated ligand–target interactions in IAI.
(a) Lagged spearman correlations (|cor| ≥0.4) between ligand and target genes across ACS samples (n=84). Ligand-target pairs have been lagged and mapped as follows before calculating the correlation: Ligand_TP1M ~ Target_TP2M; Ligand_TP2M ~ Target_TP3M; Ligand_TP3M ~ Target_TP4M [Target_TP1M and Ligand_TP4M values have not been mapped and included]. Shown target genes selected as top 2.5% of features with positive feature weight on Integrative ACS Ischemia. Shown ligands selected based on minimum regulatory potential score of 0.0012 for those targets according to the NicheNet Model (corresponding to 97% quantile of regulatory potential score). (b) Lagged spearman correlations (|cor| ≥0.4) between ligand and target genes across ACS samples (n=84). Ligand-target pairs have been lagged and mapped as follows before calculating the correlation: Ligand_TP1M ~ Target_TP2M; Ligand_TP2M ~ Target_TP3M; Ligand_TP3M ~ Target_TP4M [Target_TP1M and Ligand_TP4M values have not been mapped and included]. Shown ligand genes selected as top 2.5% of features with positive feature weight on Integrative ACS Ischemia. Shown targets selected based on a minimum regulatory potential score of 0.0012 of the selected ligands to the targets according to the NicheNet Model (corresponding to 97% quantile of regulatory potential score). (c) Exemplary visualization of lagged associations shown in circoplot in (b): Left: Violin plots showing normalized expression values of selected features for sterile ACS patients in longitudinal comparison. Timepoints were mapped as visualized by the violin plots: Ligand_TP1M ~ Target_TP2M; Ligand_TP2M ~ Target_TP3M; Ligand_TP3M ~ Target_TP4M. [Target_TP1M and Ligand_TP4M values have not been mapped and included as no corresponding ligand/target value is available, indicated by gray color]. Right: Scatter plots showing spearman correlation score of the visualized example based on mapping target molecule expression (CRP) to lagged ligand expression (IL6) as indicated by the violin plots. (d) Normalized expression values of cytokines are shown in comparison of individual timepoints of sterile ACS with CCS. Parametric distributed data were analyzed using the Ordinary One-Way ANOVA with correction for multiple comparisons by Dunnett’s test; non-parametric distributed data were analyzed using the Kruskal-Wallis test with correction for multiple comparisons by Dunn’s test. *p≤0.05, **p≤0.01, ***p≤0.001. In case only the ordinary one-way ANOVA or Kruskal–Wallis was significant, graphs are marked with a vertical bar on top. (+/- indicates the direction of the feature factor weight). Exact p-values (Supplementary Table 13) were summarized in the supplementary tables. (c) – (d) N-numbers (Supplementary Table 14) were summarized in the supplementary tables.
Extended Data Fig. 8 Gene expression changes across time and outcome in IAR.
(a) Integrative ACS Repair (Factor 4). Normalized expression values of top 1% features in longitudinal comparison visualized within heatmap and weight of the features visualized as barplot, all samples included (n=128). Divided by outcome; ‘NA’ in case no EF value has been available for the ACS samples (n=7) and for CCS and non-CCS samples. (+/- indicates the direction of the feature factor weight).
Extended Data Fig. 9 Gene expression changes across time and disease states in IC.
(a) Integrative CCS (Factor 1). Normalized expression values of top 1% features in longitudinal comparison visualized within heatmap and weight of the features visualized as barplot, all samples included (n=128). (+/- indicates the direction of the feature factor weight).
Extended Data Fig. 10 Ligand–target interactions in IC.
(a) Spearman correlations (|cor| ≥0.4) between ligand and target genes across all samples (n=128). Target genes selected as top 1% of features with negative feature weight on Integrative CCS (Factor 1). Ligands selected based on minimum regulatory potential score of 0.0012 on those targets according to the NicheNet Model (corresponding to 97% quantile of regulatory potential score). Interactions mentioned in the main text highlighted by darker color. (b) Spearman correlations (|cor| ≥0.4) between ligand and target genes across all samples (n=128). Target genes selected as top 1% of features with positive feature weight on Integrative CCS (Factor 1). Ligands selected based on minimum regulatory potential score of 0.0012 on those targets according to the NicheNet Model (corresponding to 97% quantile of regulatory potential score). Interactions mentioned in the main text highlighted by darker color.
Supplementary information
Supplementary Information
Supplementary Figs. 1–14.
Supplementary Tables
Supplementary Tables 1–16.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pekayvaz, K., Losert, C., Knottenberg, V. et al. Multiomic analyses uncover immunological signatures in acute and chronic coronary syndromes. Nat Med (2024). https://doi.org/10.1038/s41591-024-02953-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-024-02953-4
