
Abstract
Mendelian diseases contain important biological information regarding developmental effects of gene mutations that can guide drug discovery and toxicity efforts. In this review, we focus on Smith–Lemli–Opitz syndrome (SLOS), a rare Mendelian disease characterized by compound heterozygous mutations in 7-dehydrocholesterol reductase (DHCR7) resulting in severe fetal deformities. We present a compilation of SLOS-inducing DHCR7 mutations and the geographic distribution of those mutations in healthy and diseased populations. We observed that several mutations thought to be disease causing occur in healthy populations, indicating an incomplete understanding of the condition and highlighting new research opportunities. We describe the functional environment around DHCR7, including pharmacological DHCR7 inhibitors and cholesterol and vitamin D synthesis. Using PubMed, we investigated the fetal outcomes following prenatal exposure to DHCR7 modulators. First-trimester exposure to DHCR7 inhibitors resulted in outcomes similar to those of known teratogens (50 vs 48% born-healthy). DHCR7 activity should be considered during drug development and prenatal toxicity assessment.
Similar content being viewed by others
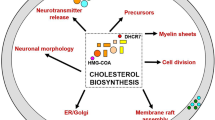

Introduction
Mendelian diseases are genetic conditions that follow a ‘traditional’ pattern of inheritance. Previously, researchers utilized information from Mendelian gene mutations to study shared underlying disease mechanisms that are common to non-Mendelian diseases in complex diseases1 and cancer.2 Mendelian diseases are also useful in studying developmental effects of gene mutations and can help researchers understand the effects of a potential pharmaceutical target or off-target effect,3 increasing the impact of their discoveries.4 Understanding the underlying mechanisms of Mendelian diseases can enable a priori prediction of fetal outcomes following prenatal pharmaceutical exposure.
In this review, we detail one orphan Mendelian disease—Smith–Lemli–Opitz syndrome (SLOS) resulting from mutations in 7-dehydrocholesterol reductase (DHCR7). These mutations affect a pathway involving vitamin D and cholesterol production. Mutations affecting vitamin metabolism can have an important role in drug response.5 In-depth study of this biological pathway enables us to explain off-target effects of prenatal drug exposure and highlights DHCR7’s importance in drug development for potential prenatal toxicity assessment.
Clinical characteristics
SLOS was first identified in 1964 when physicians described a similar pattern of congenital anomalies, including mental retardation, incomplete external genitalia and abnormalities of face, hands and feet that followed a familial inheritance pattern.6 Later, it was discovered that extremely high 7-dehydrocholesterol levels and surprisingly low serum cholesterol levels were common biomarkers of SLOS. This led to the discovery of the exact location in the cholesterol synthesis pathway that was defective in SLOS patients, namely the conversion of 7-dehyrocholesterol into cholesterol (the last step in cholesterol biosynthesis).7 Subsequently, DHCR7 was identified as the culprit gene.8 DHCR7 is the only enzyme that converts 7-dehydrocholesterol to cholesterol.9 Cholesterol cannot be produced without DHCR7.
The physical presentation of SLOS differs widely among individuals, varying by severity, genotype and other environmental factors.10 The most frequently occurring feature is 2/3 toe syndactyly (that is, ‘webbed toes’) occurring among 97% of patients followed by mental retardation with 95% of patients.10, 11 Other common signs include microcephaly (84%), postnatal growth retardation (82%), anteverted nares (78%), ptosis (70%), genital anomalies (65%) and congenital heart defects (among 54% of SLOS patients).10, 11 SLOS severity ranges across a wide spectrum. Some SLOS patients present with a mild form12 with minimal symptoms and no developmental delay.13 Others have a severe form that can result in a lack of sexual dismorphism with a functional XY karyotype and female internal and external genitalia.14
The importance of cholesterol in prenatal embryonic and fetal development, and its partial to complete absence in SLOS, helps to explain the pleotropic phenotypes within SLOS. In patients possessing homozygous null mutations in DHCR7, cholesterol production is absent and prenatal lethality results.15 Other mutations reduce DHCR7 expression to <5%, dramatically reducing cholesterol production in the body.8
Genetic characteristics
SLOS is an inherited autosomal recessive disease with each parent contributing one mutated copy of DHCR7. Inheritance follows a compound heterozygosis pattern whereby each parent contributes one copy of different mutations in DHCR7. Therefore, the SLOS patient is heterozygous for two mutations. Being heterozygous for only one mutation generally does not cause the SLOS phenotype, although instances have been reported.8, 16 Being homozygous for a null mutation in DHCR7 typically results in prenatal death.15 This explains why most full-term viable SLOS patients are compound heterozygotes. Figure 1 depicts the autosomal inheritance of SLOS in children and how compound heterozygosity is responsible for the disease phenotype. The discrepancy between the DHCR7 mutation carrier rate and SLOS incidence17 is believed to result from prenatal loss of individuals with homozygous null mutations during the first trimester.15 As in many inherited genetic conditions, de novo mutations have also been reported.18

Full-term SLOS patients are typically compound heterozygous for two distinct mutations in DHCR7 (a), whereas homozygous null individuals are detected less frequently due to prenatal lethality (b) depicts the autosomal inheritance of SLOS in children and how compound heterozygosity is responsible for the disease phenotype. Many SLOS genetic studies focus on compound heterozygous patients (a) because most homozygous phenotypes result in prenatal fatalities, reducing the detection rate (b). Both W151X and IVS8-1G>C are null mutations in DHCR7 meaning that they reduce DHCR7 expression to almost 0% in the homozygous state. Therefore, if an individual is homozygous for either of these mutations or heterozygous for the combo then little to no DHCR7 expression would result.93 On the other hand, T93M is a non-null mutation in DHCR7 that reduces DHCR7 expression by 5% when compared to normal.37 Therefore, a compound heterozygous patient with one IVS8-1G>C null mutation and one T93M mutation would have around 45% functional DHCR7 and SLOS would result, but prenatal fatality would be averted (a). DHCR7, 7-dehydrocholesterol reductase; SLOS, Smith–Lemli–Opitz syndrome.
Importantly, the relationship between null mutations in DHCR7 and SLOS severity is not one-to-one because variations in the maternal genome can increase the amount of cholesterol passed by the placenta to the fetus15 modulating the offspring’s phenotype. Because cholesterol is critical during early development, having increased prenatal cholesterol levels distributed from the mother via the placenta can mitigate many SLOS symptoms.15 Diverse factors modulate SLOS severity, therefore it is not possible to completely predict the disease phenotype using genotype information alone or vice-versa.19
Compendium containing SLOS-inducing DHCR7 mutations
Development of the DHCR7 SLOS mutation compendium
SLOS patients are compound heterozygotes for diverse mutations in DHCR7 or homozygous for non-null mutations. SLOS is a rare disease (~1 in 40 000) and studies typically involve only small cohorts of patients. Even with small cohorts, many DHCR7 mutations have been reported. Therefore, we reviewed the literature and developed a compendium of SLOS-inducing DHCR7 mutations. We collected the reported frequencies for each mutation across all studies, and when available, we extracted the patient genotype (that is, compound heterozygous mutant alleles) and their geographic location or ethnicity. We are interested in the ethnicities of SLOS patients and their corresponding genotypes because certain gene variants affect outcomes only within a given ethnicity (for example, ACBCB1 variant in Caucasians).20
To develop our compendium, we analyzed all publically available DHCR7 mutations contained in the Human Gene Mutation Database (HGMD)21, 22 and their corresponding publications (HGMD last accessed in May 2015). We kept track of patients’ DHCR7 genotype information and their ethnicity/geographic origin when available (described later). The aggregated allele frequencies for all DHCR7 mutations across 30 studies are given in Supplementary Table S1. Our review focuses only on deleterious mutations to DHCR7, and therefore does not include 13 silent DHCR7 mutations.23 Using the HGMD, we found 138 distinct publically available mutations to DHCR7 and 165 reported (with the additional 27 mutations being proprietary, and only accessible via paid membership).
Our literature review revealed additional DHCR7 mutations (by investigating papers cited in those retrieved papers) and overall our compendium contains 147 DHCR7 mutations, 145 of these being SLOS-inducing mutations. One missense mutation, W158C, was found in an unaffected sibling of a SLOS individual (noted in Supplementary Table S1), and another mutation, G344D, was reported in a patient with holoprosencephaly (noted in Supplementary Table S1) a different mutation at the same position, G344R, was reported in a SLOS patient. Mutations in DHCR7 that result in holoprosencephaly were also included in the compendium and denoted in Supplementary Table S1.
Common SLOS-inducing DHCR7 mutations
We found that 12 mutations (of 145 SLOS-inducing mutations) occurred with allele frequency of at least 1% across the 30 studies we reviewed (Table 1). The most common of these were IVS8-1G>C (28.446%), T93M (9.384%), W151X (8.407%) V326L (5.083%), R404C (3.519%) and R352W (3.324%). We compared this result to data obtained from 60 706 assumed healthy individuals (that is, non-SLOS) to determine the frequency of various DHCR7 mutations in that reference population. We used the ExAC database24 available at http://exac.broadinstitute.org/ (accessed November 2015). These results are also given in Table 1. We believe the ExAC population are non-SLOS individuals, however it is likely that they have other conditions. Therefore, from henceforth we refer to this population as the ExAC population. The majority of frequent DHCR7 mutations in SLOS patients also occur at higher frequencies in the ExAC population (this is intuitive). There are two exceptions: T93M that was common among SLOS patients (9.4%) but rare in the healthy population (only 3 allele mutations observed out of 60 706 individuals). Another counter-intuitive result was that R242C was found relatively frequent in the ExAC population (10 allele mutations observed), but was only mutated in 1.2% for SLOS. We present the top 10 DHCR7 SLOS-inducing mutations as ranked by their incidence in the ExAC population and their corresponding incidence in our SLOS patient set in Table 2. All of the SLOS DHCR7 mutations were <1% frequency in the ExAC population. We compared our entire compendium of DHCR7 mutations to ExAC. The results of this comparison including accession numbers and chromosomal locations are given in Supplementary Table S2.
A SLOS review from 2005, mentioned 11 mutations with at least 1% frequency across SLOS patients.25 We compared our findings to theirs and noticed that the top 8 mutations remain unchanged. We noted that two additional mutations were on our list, namely R352Q (2.151%) and P51S (1.173%) and one mutation on their list was not included in ours, namely S169L (Yu: 1.6%) having a frequency of 0.868% in our compilation of 30 studies (Supplementary Table S1; Table 1).
The availability of Korean genetic data resulted in the addition of R352Q26 to the list of frequent SLOS mutations (⩾1%). Importantly, although SLOS occurs more frequently among Europeans,17, 27 mutations in DHCR7 exhibit an evolutionary pressure towards Northern Europeans and Northeast Asians,28 indicating that SLOS may be currently under-reported among Northeast Asians. Therefore, as more genotype data becomes available from non-European SLOS patients, we would expect the common allele frequencies to change somewhat and become less biased towards Northern Europeans.
A couple of DHCR7 mutations were reported to cause SLOS in the heterozygous form.8, 16, 29, 30 However, it is likely that an unidentified suspected variant was present in these instances, as SLOS results from two defective copies of DHCR7, either in a compound heterozygous state (2 different DHCR7 mutations) or a homozygous mutated state. Importantly some asymptomatic siblings of SLOS patients were heterozygous carriers of the null W151X mutation (they should have around 50% expression of DHCR7) and they exhibited no symptoms of SLOS suggesting that the disease causing state requires DHCR7 expression to be <50%.8 This also confirms that one copy of a DHCR7 mutation does not result in SLOS.
DHCR7 mutations vary by geographical location
Specific DHCR7 mutations exhibit geographic dependency
SLOS incidence ranges by geographical location and ethnicity because carrier frequencies vary by region.17 Reports range from 1 in 9000 from Czechoslovakia and Central European populations to 1 in 40 000–70 000 in Canada.17, 31 The incidence in both the United States of America (USA) and Europeans is 1 in 30 000 live births.17, 27 Variations in reported incidence rates are thought to be due to sampling bias differences. The carrier rate, or the proportion of individuals with one copy of a known SLOS-inducing DHCR7 mutation, is estimated to be 3% among persons of European ancestry, 27 whereas another study found 1–2% among Caucasians.32 In the USA the carrier frequency is 1%,33, 34 although in the state of Utah the carrier rate is 4%.35
Mutation frequencies are known to vary by geographic location/ethnicity.36 Likewise, the spectrum of DHCR7 mutations varies by geographical location. There is evidence that T93M is the founder mutation with origins in the Mediterranean basin37 and is common in Italy, Spain38, 39, 40 and Portugal.41 On the other hand, Northern Europeans (Austrians, Germans, Dutch) present with the W151X mutation more frequently.15, 38 In Korea, three mutations account for most of the observed variants.26 Persons of African ancestry rarely develop SLOS, however some carriers were observed among persons of African ancestry.27 One SLOS patient with Spanish-African mixed ancestry was identified, this patient had T93M (the common Spanish SLOS allele) and V281M.37 The carrier frequency of DHCR7 mutations among African Canadians was low at 0.79%.33
The relationship between the specific combinations of DHCR7 mutations and ethnicity/geographic location of SLOS patients warranted further exploration. Therefore, we extracted all genotype data for SLOS patients that also contained ethnicity details, or where the ethnicity could be deduced (for example, a German study enrolling patients in Germany). Not all studies listed in Supplementary Table S1 reported the genotype and ethnicity information for DHCR7 mutations. The aggregated results are shown in Table 3 with genotype data from 21 studies representing 229 patients from 26 different countries/ethnicities (if mixed ethnicities are included). The exon distribution of the 1024 alleles with DHCR7 mutation information available is shown in Figure 2a. The proportion of DHCR7 mutations that exist in a trans-membrane domain are shown in Figure 2b. Three protein structure models are shown in Figure 2b because the 3-dimensional structure of DHCR7 has not been solved yet. The three models are: the human protein reference database containing trans-membrane exon bound information, the Waterham and Wanders's study42 and the Fitky et al. study.8 We also analyzed the 170 compound heterozygous patients (typical SLOS patients) with geographic/ethnicity data from Table 3 as shown in Figures 2c and d; Supplementary Figure S1.

Certain exons and functional regions are enriched for SLOS-inducing mutations in DHCR7 and mutation spectrum varies by region and ethnicity. The overall exon distribution of alleles is shown in (a) for all 1024 alleles. Notice that the intronic null mutation (renders exon 9 non-existent) is the most common followed by mutations in exon 9. Exons 4 and 6 also feature prominently in SLOS-inducing mutations. (b) depicts the proportion of distinct DHCR7 mutations (left) and overall mutations (right) that occur in the trans-membrane region of the protein. The 3-dimensional protein structure for DHCR7 has yet to be published. However, three different models have been described indicating transmembrane domains including the Human Protein Reference Database, Waterham and Wanders42 and Fitzky et al. 8 Note that the Waterham model42 results in the largest proportion (62.109%) of SLOS-inducing mutations being flagged as occurring in the transmembrane domain. (c) The allele frequency distribution for patients with ethnicity or country of origin information (170 patients had this information available). Some ethnicities only had one patient including, Ashkenazi, Turkish and Spanish/African (a patient with mixed ancestry). (d) The exon locations for each compound heterozygote pair (each patient has 2 mutations in DHCR7). Note that different ethnicities or countries of origin have different mutation patterns. For example Asian patients frequently have one mutation in exon 8 and one mutation in exon 9; whereas patients from Europe (unspecified lower left-hand corner of (d) or Northern Europe tend to have one mutation in exon 6 and other in exon 9. Patients from Southern Europe frequently had two different mutations in exon 9 or one mutation in exon 4 and one in exon 9. Ethnicity groupings: Europe (European but not otherwise specified and N-S Europeans); N. Europe (Dutch, Hungarian, Polish, German, Austrian, United Kingdom, Irish); S. Europe (French, Italian, Spanish, Portuguese, Greek); S. America (Brazil, United States of America-Hispanic); Asia (Korean, Japanese, Japanese-Dutch). DHCR7, 7-dehydrocholesterol reductase; SLOS, Smith–Lemli–Opitz syndrome.
Exon ‘Hotspots’ distinguish among continents and between north and south
Figure 2c illustrates how specific DHCR7 mutations occur commonly among certain ethnicities. For example, R352Q and G303R occur almost exclusively among Asian populations (Asian: Korean, Japanese, Japanese-Dutch). Other mutations, for example, G147D, occur among Southern Europeans (S. Europe: French, Italian, Spanish, Portuguese, Greek). There are some distinctions between Northern and Southern Europeans in terms of DHCR7 mutations. For instance, the null mutation IVS8-1G>C occurs more frequently among Southern Europeans, whereas the null mutation W151X occurs more frequently in Northern Europeans. We also found exon ‘hotspots’ for the pairs of SLOS-inducing DHCR7 mutations. The combination of exons 4 and 9, and also 6 and 9 tended to occur frequently across the whole cohort (see ‘All’ in Figure 2d). When we stratified by ethnic group, we found that mutations in exons 4 and 9 or the exon 4–9 ‘hotspot’ were more frequent among Southern Europeans, whereas the exon 6-9 hotspot occurred frequently in Northern Europeans. We observed marked differences for the Asian population with the exon 8–9 hotspot occurring more frequently. Individuals from South America (S. America: Brazil, United States of America-Hispanic) had three patterns that were equally frequent, the exon 4–9 hotspot (also common among S. Europeans), the exon 7–9 hotspot (unique to S. Americans), and the exon 8–9 hotspot (common among Asians). Importantly, the frequent N. European hotspot (exon 6–9 hotspot) was absent from the S. American result.
A clear relationship exists between geography/ethnicity and specific DHCR7 mutations as shown in Figure 2d and has been described previously.26, 37, 43, 44 In addition, we demonstrate that the specific exon ‘hotspots’ also vary by geographic location/ethnicity (Figure 2d).
Pathway links DHCR7, vitamin D synthesis and cholesterol synthesis
Evolutionary advantage for DHCR7 mutations
Heterozygote advantage among carriers has been proposed previously17 based on the relationship between DHCR7 and vitamin D. Heterozygote carriers have lower amounts of functional DHCR7 resulting from their carrier state.8 This would lower the amount of available 7-dehydrocholesterol that is converted to cholesterol. Subsequently, levels of 7-dehydrocholesterol would increase allowing more available 7-dehydrocholesterol to be converted to vitamin D upon exposure to ultraviolet B light. This would protect individuals from developing rickets (a vitamin D deficiency condition that results in bone softening and malformations) and osteomalacia (an adult form of rickets).17
Certain DHCR7 variants were found to be evolutionarily favored in Northern climates, including Europe and Asia28 suggesting a selective pressure and a plausible heterozygote advantage for those variants. Researchers found certain DHCR7 variants were associated with lower vitamin D levels in the general population45 and among individuals with polycystic ovary syndrome.46 This further links vitamin D levels with DHCR7 variants.
Historically, there would have been an evolutionary pressure to maximize the small amount of sunlight available for vitamin D production in Northern climates due to lower sunlight exposure. In addition, benefits from prenatal vitamin D supplementation are also modulated by geographic location and other factors.47 Therefore biological mechanisms that maximize vitamin D production and absorption would be selected for in the North. A heterozygote advantage for individuals with one DHCR7 mutation would therefore exist to allow most of the body’s 7-dehydrocholesterol to be converted into vitamin D (and not cholesterol). This has been used to explain why Northern populations have a higher prevalence of SLOS.11
Carriers also are hypothesized to have a reproductive advantage because of the reduced fetal death due to rachitic cephalopelvic disproportion.11, 35 This is again related to increased vitamin D production causing improved bone formation. Improved hip formation is thought to enable females to produce more offspring and thereby provide another carrier advantage.11, 35 In addition, vitamin D has been shown to increase chances of pregnancy for infertile women receiving in vitro fertilization.48 Vitamin D has many pleiotropic roles in reproductive outcomes and can affect both male and female fertility.49 Therefore, there could be multiple mechanisms that could explain why evolution favors vitamin D production in Northern climates through mutations in DHCR7 (and other genes).
Biological mechanism: DHCR7’s effect on vitamin D and cholesterol synthesis
The biological mechanism that connects DHCR7 mutations (SLOS carriers) and Northern climate involves regulation of 7-dehydrocholesterol, vitamin D and cholesterol. Typically, 7-dehydrocholesterol can be converted into either cholesterol or vitamin D (cholecalciferol). The conversion of 7-dehydrocholesterol to vitamin D occurs in the skin upon exposure to ultraviolet B light (290–320 nm).50 Under normal sunlight conditions, only around 15% of available 7-dehydrocholesterol will be converted to vitamin D, whereas excess 7-dehydrocholesterol in the skin will be converted to inert compounds for degradation.51, 52 DHCR7 is the sole enzyme used to convert 7-dehydrocholesterol to cholesterol.53 A shortage of DHCR7 would decrease the body’s ability to convert 7-dehydrocholesterol into cholesterol thereby causing a build-up of 7-dehydrocholesterol, which could be converted into vitamin D in the skin. Each piece of this mechanistic pathway is depicted in Figure 3 along with the corresponding literature references.

Literature-derived pathway illustrates how 7-dehydrocholesterol reductase effects vitamin D production by removing 7-dehydrocholesterol and the effects of drugs on this pathway. Drugs that enhance DHCR7 function result in reduction in vitamin D production. This occurs because DHCR7 causes more 7-dehydrocholesterol to be converted to cholesterol. This indirectly inhibits vitamin D production by reducing the amount of 7-dehydrocholesterol that can be converted into cholecalciferol in the skin. Homozygous null mutations in DHCR7 completely inhibit functionality and result in patients with SLOS with heterozygous patients showing increased protection against rickets and osteomalacia. The dashed line indicates a feedback inhibition loop. 1 (ref. 53), 2 (ref. 45), 3 (ref. 94), 4 (ref. 7), 5 (ref. 57), 6 (ref. 58), 7 (ref. 59), 8 (ref. 54), 9 (ref. 28), 10 (ref. 50). DHCR7, 7-dehydrocholesterol reductase; SLOS, Smith–Lemli–Opitz syndrome.
An indirect negative feedback loop, established in laboratory studies, allows vitamin D levels to regulate DHCR7 activity and prevent hypervitaminosis D (that is, toxically high vitamin D levels).54 SLOS patients have high 7-dehydrocholesterol levels. Therefore, from Figure 3 and the literature, we would expect them to have high vitamin D levels. However, vitamin D levels in SLOS patients are reported to be low.55 This could be due to many lifestyle factors that occur when a patient is seriously ill with SLOS. One possible biological explanation that fits with the literature-derived mechanism (Figure 3) is that SLOS patients spent less time outdoors thereby reducing their ultraviolet B exposure (another critical requirement for vitamin D synthesis).55
Genetic understanding of SLOS-inducing DHCR7 mutations: implications for future work
Changing our understanding of SLOS genetics using large-scale genomics studies
Using ExAC,24 we were able to compare the frequencies of DHCR7 SLOS-inducing mutations in the SLOS population (extracted from the literature) and the assumed-healthy ExAC population. We found that there were many differences. To illustrate some of these differences, we plotted the overall incidence of DHCR7 mutations in SLOS against the incidence in the healthy population (ordered by healthy population). Several DHCR7 mutations are much more frequent among SLOS individuals then expected given the background population rate (Figure 4). For example, T93M is the second most frequent DHCR7 mutation in SLOS patients, but it is comparatively rare among the ExAC population (Figure 4).24 This could be due to sampling bias differences between the SLOS cohort and the ExAC population. For example, T93M is thought to be the founder SLOS mutation and is common in Italy, Spain and Portugal.37, 38, 39, 40, 41 The ExAC population may be under-represented for Southern Europeans. In ExAC they distinguish a ‘Finnish’ European cohort from a non-Finnish European cohorot (representing Northern American peoples and other Europeans), but it is unclear how many Southern Europeans are represented in that cohort. Therefore, some of these differences are likely due to sampling bias.

DHCR7 SLOS-inducing mutation frequency in SLOS patients vs frequency from ExAC population. Some mutations occurred more frequently in the SLOS population given their frequency in the healthy population (for example, T93M, V326L, R404C, R352W). A possible explanation for this could be that these mutations occur in certain ethnic populations and those populations are under-represented in the ExAC population. DHCR7, 7-dehydrocholesterol reductase; SLOS, Smith–Lemli–Opitz syndrome.
In addition, we were able to identify DHCR7 mutations that were predicted to be damaging using ExAC. This revealed 24 mutations (Table 4). Eight of those mutations were already known to be implicated in SLOS and were contained in our compendium. Six more mutations occurred at the same amino acid position of another mutation that has been implicated in SLOS (demonstrating the importance of that amino acid position in SLOS). However, 12 mutations were never reported as being found in SLOS patients. There are two potential reasons for this: (1) the mutation is not disease causing (this is difficult to ascertain without the protein structure); and (2) the mutation only exists in an under-studied research population. For example, one mutation (R207X) was found to have 2.2% frequency among the healthy African population from ExAC. This means that R207X is polymorphic among Africans. However, this mutation has never been implicated in SLOS. Its possible that Africans are not often diagnosed with SLOS and then sequenced because they often come from resource-poor environments. The 12 mutations in Table 4 that have never been implicated in SLOS are worthy of further investigation by researchers to ascertain whether they are deleterious. If these mutations are deleterious, the next research question would be why African populations are under-diagnosed for SLOS.
Implications for causality
Hill56 established a set of nine criteria for determining whether an association was causal or not. We will focus our discussion on three of these criteria: strength, plausibility and coherence. Strength is difficult to assess in SLOS because the DHCR7 mutations are often only found among SLOS patients (and researchers are biased to look specifically for mutations in DHCR7). As we can see from Table 2 the incidence of these mutations in the ExAC population is very low. This helps to strengthen our belief in the relationship between DHCR7 mutations and SLOS, especially for mutations that are disproportionately high among SLOS patients (for example, T93M in Figure 4). However, the fact that some mutations are very high among SLOS patients and very low among the ExAC population could be due to sampling bias between the two populations. Some DHCR7 mutations were found in the ExAC population that is predicted to be functionally deleterious. However, not all of these have been reported to be SLOS-Inducing. This could be due to ethnic differences between the two datasets (that is, certain groups are under-studied and therefore some of the predicted damaging mutations are actually un-reported SLOS-Inducing mutations) or it could be because some predicted damaging mutations are not functionally damaging (this would be easier to determine if the structure of DHCR7 were known). Because the protein structure of DHCR7 remains unknown it is difficult to predict for certain whether a mutation is deleterious or not.
A paper by Lanthaler et al.15 found that certain maternal genetic signatures could help rescue the SLOS phenotype by increasing the amount of cholesterol passed to the offspring via the placenta. This variation in the maternal genome could perhaps explain some of the discrepancies between our literature-derived SLOS compendium and the ExAC population. For example, in addition to the requirement that two DHCR7 mutations be inherited, it may also be necessary to have a certain placenta state (or placental gene mutation) to acquire the disease. This could perhaps explain why some common DHCR7 mutations from ExAC are not found to be SLOS-inducing (even though they are predicted to be damaging). However, it is too early to determine yet because of sampling bias issues and under-reporting among certain ethnicities. Further investigation could help provide additional coherence to reported DHCR7 mutations in the literature. Therefore, this remains an open area of research.
Pharmacological effects of DHCR7 modulators
Importantly, pharmaceutical drugs have been shown to modulate DHCR7 activity in various ways. Several anti-psychotic drugs were found to enhance DHCR7 activity.57, 58, 59 Other drugs, not otherwise known to modulate DHCR7, can cause high 7-dehydrocholesterol in the absence of SLOS.60 Currently, pharmaceuticals are being designed to inhibit DHCR7 as a suggested treatment for hepatitis C.61 However, diverse therapeutic uses exist for targeting DHCR7. In this section, we review all known pharmaceuticals, compounds, chemicals, and toxins known to modulate DHCR7.
DHCR7 modulators
Expression
We used the Comparative Toxicogenomics Database (CTD)62, 63 to retrieve articles describing compounds that modulate DHCR7 expression (data retrieved February 2015). We then manually reviewed the resulting literature references and only retained compounds with direct evidence of modulating DHCR7 expression in studies using human tissue. Table 5 contains the list of modulators with corresponding references. Several other compounds were found in CTD to modulate DHCR7, but failed our requirements listed above these are presented as potential DHCR7 modulators in Supplementary Table S3. Overall, we found five pharmaceuticals and two inorganic compounds increased DHCR7 expression and three toxins decreased DHCR7 expression. Antipsychotic drugs, such as haloperidol, clozapine and chlorpromazine, are known to alter expression in cardiovascular genes64 in addition to DHCR7 (increased DHCR7 activity would increase the rate of cholesterol synthesis, Figure 3). Details on the three toxins are described in the Supplementary Material.
Inhibition
We used ChEMBL, a freely-available semi-curated database of bioactive molecules,65 to find all pharmacological compounds that directly inhibit DHCR7 (Table 5). This includes approved chemotherapeutics, and investigational chemotherapeutics either in clinical or preclinical trials. When available we report IC50 values (Table 5). Drugs with IC50 values <10 000 nm or 10 μm are considered potentially pharmacologically relevant inhibitors.66, 67 We included tamoxifen (IC50=12 nm) and doxorubicin (IC50=150 nm) as FDA-approved DHCR7 inhibitors.
DHCR7 inhibitors are used to treat cancer, typically breast cancer. For example, nafoxidine inhibits DHCR7 (ref. 68) and is a known estrogen antagonist. Several clinical trials were performed in the 1970s for nafoxidine.69, 70, 71, 72 The major reaction to the drug consisted of dermatitis (55% of patients) exacerbated by sunlight.72 This indicates that the drug effected patients’ dermal response to sunlight exposure (which could be related to its inhibition of DHCR7 and dysregulation of vitamin D synthesis pathways).
Summary modulators
Overall, we found that five approved pharmaceuticals increase DHCR7 expression, whereas two inhibit DHCR7. Two inorganic compounds increase DHCR7 expression, whereas three toxins (including a metabolite of arsenic) decrease DHCR7 expression and one toxin decreases protein activity. Ten pharmaceutical inhibitors of DHCR7 are in various stages of clinical development. This includes seven inhibitors in the pre-clinical stage and three inhibitors in the clinical trials stage (Supplementary Table S4). DHCR7 Inhibitors are used or will be used (if approved) for cancer treatment.
Investigating fetal outcomes following prenatal exposure to DHCR7 modulators
Overview
The biological mechanisms underlying pharmacological teratogenicity and adverse fetal outcomes are complex involving many genetic and environmental factors. In the United States of America, developmental defects occur in ~3–5% of live-born children.73 Among these defects between 2 and 3% are classifiable as teratogen-induced meaning that the defect occurred due to a known or suspected prenatal environmental exposure.73 It is further estimated that <1% of teratogenic effects have a pharmacological origin.74
Interactions between pharmaceutical drugs and genetics could result in increased teratogenicity among certain individuals. For example, holoprosencephaly clusters in families,75 can result from mutations in DHCR7,76 and can also occur after exposure to certain drugs, namely lovastatin.77, 78 It is reasonable to assume that various combinations of those factors (for example, drug exposure, DHCR7 mutations, familial risk factors) could result in increased likelihood for teratogenic effects. However, for the purposes of this review, we focus on fetal outcomes resulting from prenatal drug exposure to DHCR7 modulators.
Because of the importance of functioning DHCR7 during fetal development, we decided to investigate the outcomes of prenatal exposure to two approved pharmaceutical DHCR7 inhibitors (Supplementary Table S4) and compare them against five approved pharmaceutical drugs that increase the DHCR7 expression. We were interested in the fetal outcomes of both DHCR7 inhibitors and those that increase expression to determine if DHCR7 inhibitors resulted in increased detrimental fetal outcomes. We made this hypothesis because DHCR7 inhibition results in a pharmaceutically induced SLOS-like fetal development environment and therefore prenatal exposure to these inhibitors should result in SLOS like adverse outcomes.
PubMed literature review on fetal toxicity
Using PubMed, we retrieved all relevant observational studies on fetal outcomes due to prenatal exposure of each drug. The full details regarding the semi-automated query and retrieval process for included articles are described in the Supplementary Material. We reviewed each observational study carefully recording the number of pregnancy outcomes per drug treatment. Seven main pregnancy outcomes were used: born-healthy, spontaneous abortion (or miscarriage), elective termination (or induced abortion), neonatal death (died within 1st week of birth), still birth (or intrauterine death), ectopic pregnancy and major/minor fetal malformations or defects or congenital anomalies. Whenever possible we included information regarding the trimester of drug exposure, as this is critical information for understanding teratogenic effects, realizing that SLOS-like symptoms would result mainly from first-trimester exposure (when cholesterol production is critical for proper structural formation). As controls, we included one known teratogen in our search and one known pregnancy-safe drug. We selected isotretinoin (tradename: accutane) as our known teratogen and levothyroxine as our known pregnancy-safe drug (that is, FDA category A).
Our method uses data gleaned from literature reports on fetal outcomes following prenatal drug exposure. Therefore, this data suffers from reporting bias. This occurs because reports on a drug’s effects may be more likely if the effect is deleterious or severe. To address this issue, we included two ‘control’ drugs and followed the same procedure (that is, literature review and extraction of outcomes). One ‘control’ drug was a known pregnancy safe drug—levothyroxine and the other ‘control’ drug was a known teratogenic drug—isotretinoin. We can compare our findings of drugs that modulate DHCR7 to these two ‘controls’ to determine if any effect we observe is significant and worthy of further investigation.
A certain number of adverse fetal outcomes occur in the general population. For additional comparison purposes, we also included data from the Centers of Disease Prevention and Control (CDC) on pregnancy outcomes (that is, live born, spontaneous abortion, elective termination) collected across all races from both 1990 and 2008 for reference.79 We coupled this with data stating that 3% of live-born babies (1 in 33) have a birth defect.80 Table 6 displays our findings along with their corresponding references.
Fetal outcomes from prenatal exposure to DHCR7 modulators
The overall results for each of the seven FDA approved DHCR7-modulating pharmaceuticals are given in Figure 5. For comparison purposes, known pregnancy-safe levothyroxine and known teratogen isotretinoin (or accutane) are also included. The breakdown of these drugs across the seven pregnancy outcomes is given in Figure 5a. The number of deformations (possible teratogenic effects) vs number of healthy babies is shown in Figure 5b. Drugs that inhibit DHCR7 resulted in deformities among 10.9% of babies born, whereas drugs that increase DHCR7 expression resulted in deformities among 5.5%. This can be compared to the CDC background of 3.0% and levothyroxine’s rate of 0.0% (the known pregnancy-safe drug). Although DHCR7 inhibitors result in increased risk of deformities, the rate was still much lower than the well-known teratogen—accutane or isotretinoin—at 47.7%. We also statistically compared how DHCR7 activity affected fetal outcomes. Using the CDC background rate from 2008 (http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_07.pdf)79 (accessed in January 2016), we performed a Fisher’s exact test for healthy vs non-healthy (this includes born with birth defect/deformity, spontaneous abortion, elective terminations). As expected, levothyroxine (pregnancy safe drug) was not statistically different from the background rate (P-value=0.1974). We found that DHCR7 inhibitors were highly enriched for adverse fetal outcomes (odd ratio=6.0, P-value<0.001) with DHCR7 promoters showing less enrichment (odd ratio=3.3, P-value<0.001). Neither came close to the known teratogen isotretinoin (odd ratio=34.8, P-value<0.001).

Fetal outcomes of prenatal exposure to DHCR7 modulators compared to CDC prevalence and a known teratogenic drug (i.e., isotretinoin or accutane) and a known pregnancy safe drug (that is, levothyroxine). (a) contains aggregated results across all trimesters for regarding pregnancy outcomes. (b) Only born-healthy and born with fetal malformations, defects or congenital anomalies. Therefore, we excluded spontaneous abortions, elective terminations, stillbirths, ectopic pregnancies and neonatal deaths. The CDC provides data on live-born babies, elective terminations and fetal losses. We report fetal losses as spontaneous abortions in (a). We also split the number of live-born babies into born with defects and born-healthy using the statistic that 3% of live-born babies has a congenital anomaly. Notice that in (a) the number of fetal malformations/anomalies is higher among DHCR7 modulators including those that increase expression of DHCR7 (5.5%). However, drugs that inhibit DHCR7 have an increase in fetal malformations (10.9%). None of these levels comes close to the known teratogen, isotretinoin or accutane, with 47.7% born with malformations (out of total born). Many patients that are pregnant elect to terminate their pregnancy (a), which may be due to detect anomalies, however data on malformations among aborted fetuses is typically not available. We took all reported results where first-trimester exposure occurred (even if exposure persisted throughout the pregnancy) or where the exposure was listed as ‘early pregnancy’ as this appeared to indicate first-trimester exposure and these are shown in (c). Many known first-trimester accutane or isotretinoin resulted in elective terminations or induced abortions. Therefore, we included elective terminations and spontaneous abortions in (d) along with live births (healthy or malformed).
We also investigated the relationship between first-trimester exposure to DHCR7 modulators and increased risk of teratogenic effects (Figures 5c and d). We were especially interested in first-trimester effects because reduction in cholesterol has been shown to cause severe teratogenic effects when the exposure occurs during the first-trimester.78 By inhibiting DHCR7 during the first-trimester, cholesterol production is also inhibited, therefore, we would expect similarly severe teratogenic effects.78 In addition, studies have shown that exposure to chemotherapeutic drugs, for example, doxorubicin and tamoxifen, during the first-trimester is associated with increased risk of complications to the fetus81 then exposure during other trimesters. First-trimester results for all pregnancy outcomes are shown in Figure 5c. We grouped drugs by their DHCR7 effect given in Figure 5d. Counts and percentages of pregnancy outcomes for first-trimester exposure to DHCR7 modulating drugs are provided in Table 7.
We found that first-trimester exposure to drugs that increase DHCR7 expression was comparable to first-trimester exposure of a known pregnancy-safe drug with 73.5% born-healthy among drugs increasing DHCR7 expression vs 72.7% for levothyroxine. Even more importantly, we found that first-trimester exposure to DHCR7 inhibitors was comparable to a known teratogen (that is, isotretinoin) with 50% born-healthy among drugs that inhibit DHCR7 vs 48.3% for isotretinoin. However, the sample size for first-trimester exposures was small (N=40 for known DHCR7 inhibitors) indicating that caution must be taken when interpreting these results.
Interpretation of fetal results
We found that tamoxifen has an IC50 of 12 nm indicating that it inhibits DHCR7 at a pharmacologically potent level.66, 67 We also found that tamoxifen exposure (especially during 1st trimester) resulted in increased fetal malformation risk. When compared to tamoxifen, doxorubicin had a lower ability to inhibit DHCR7 (IC50=150–10 000 nm) and likewise a lower risk of adverse fetal outcomes.
In total, five pregnancies resulted in fetal malformations/defects or congenital anomalies from first-trimester tamoxifen exposure while two pregnancies resulted from first-trimester doxorubicin exposure. The anomalies resulting from first-trimester tamoxifen exposure appeared to have phenotypes similar to SLOS. These included, two cases of genitalia deformities including ambiguous genitalia,82, 83, 84 cleft palate (a form of holoprosencephaly)84, 85 and Goldenhar’s syndrome, which is characterized by facial deformities83, 86 and one hand deformation.84 Note that the facial deformities and cleft palate (a form of holoprosencephaly) can occur in SLOS patients. Genital anomalies occur frequently among SLOS patients.10, 11, 14 Hand deformations are rare among SLOS patients, however toe anomalies (for example, toe syndactyly) are common.10, 11
Two deformed babies reported in the literature were born after first-trimester doxorubicin exposure. One had genitalia issues,87 whereas the other had hydrocephalus.88 These congenital defects are highly related to the pleiotropic effects of SLOS (and defective functioning of DHCR7) indicating the distinct possibility that pharmacological inhibition of DHCR7 during the first-trimester of pregnancy results in teratogenic effects similar to the physical manifestations of SLOS.
Comment on vitamin D3’s in-direct inhibition of DHCR7
Of note, vitamin D (cholecalciferol) decreases the DHCR7 activity through an indirect mechanism.54 There has been some speculation on the possibility of teratogenic affects resulting from high doses of vitamin D when taken as a prenatal supplement.89 However, a recent review of vitamin D supplementation among pregnant women found that vitamin D supplementation may reduce the risk of pre-eclampsia, low birth weight, preterm birth90, 91 and adverse kidney outcomes in offspring.92 Because the mechanism of vitamin D3’s inhibition of DHCR7 is indirect (unlike our studied pharmacological DHCR7 inhibitors) it is likely to be mediated through a complex pathway with many interacting feedback loops. Studies involving moderate prenatal vitamin D supplementation have reported no adverse effects.89 Therefore, we would not expect vitamin D to result in SLOS-related teratogenic effects, although additional studies would be needed to ascertain the effects of large doses of prenatal vitamin D supplementation.
Future directions and conclusion
In this review, we demonstrate the utility of an in-depth exploration of one Mendelian orphan disease: SLOS. We contribute a compendium of SLOS-inducing DHCR7 mutations, their incidence and geographic distribution. We also include the incidence of these mutations in a large population from ExAC. This helped us illustrate important differences between mutation frequencies in SLOS vs an assumed healthy population. Comparing our SLOS mutation compendium to ExAC allele frequencies allowed us to raise some important research questions both for studying under-represented ethnic groups, such as Africans (found to exhibit theoretically damaging DHCR7 mutations in ExAC), and for understanding genetic drift of DHCR7 mutations.
We went one step further and explored the prenatal effects of pharmaceuticals that target DHCR7. We posited that pharmaceutically induced DHCR7 inhibition would result in fetal outcomes similar to those seen in SLOS (miscarriage, fetal malformations, ambiguous genitalia). We reviewed the literature for observational studies on fetal outcomes due to drug exposure. We found that exposure to DHCR7 inhibitors during the first-trimester of pregnancy resulted in fetal deformities/malformations or anomalies in 50% of conceptions born-healthy vs 48% for a known teratogen control. Contrastingly, 73% were born-healthy to those on drugs that increased DHCR7 expression compared to 73% for a known pregnancy-safe control. These results indicate that screening for DHCR7 inhibition during the pre-clinical phase of drug toxicity may be helpful in identifying drugs with potential to induce adverse fetal outcomes before the drug is released to the market.
References
Blair David R, Lyttle Christopher S, Mortensen Jonathan M, Bearden Charles F, Jensen Anders B, Khiabanian H et al. A nondegenerate code of deleterious variants in mendelian loci contributes to complex disease risk. Cell 2013; 155: 70–80.
Melamed RD, Emmett KJ, Madubata C, Rzhetsky A, Rabadan R . Genetic similarity between cancers and comorbid Mendelian diseases identifies candidate driver genes. Nat Commun 2015; 6: 7033.
Bunnage ME, Gilbert AM, Jones LH, Hett EC . Know your target, know your molecule. Nat Chem Biol 2015; 11: 368–372.
Fishman MC, Porter JA . Pharmaceuticals: a new grammar for drug discovery. Nature 2005; 437: 491–493.
Carr D, Whiteley G, Alfirevic A, Pirmohamed M . Investigation of inter-individual variability of the one-carbon folate pathway: a bioinformatic and genetic review. Pharmacogenomics J 2009; 9: 291–305.
Smith DW, Lemli L, Opitz JM . A newly recognized syndrome of multiple congenital anomalies. J Pediatr 1964; 64: 210–217.
Tint GS, Irons M, Elias ER, Batta AK, Frieden R, Chen TS et al. Defective cholesterol biosynthesis associated with the Smith–Lemli–Opitz syndrome. N Engl J Med 1994; 330: 107–113.
Fitzky BU, Witsch-Baumgartner M, Erdel M, Lee JN, Paik Y-K, Glossmann H et al. Mutations in the Δ7-sterol reductase gene in patients with the Smith–Lemli–Opitz syndrome. Proc Natl Acad Sci USA 1998; 95: 8181–8186.
Wilcox C, Feddes G, Willett-Brozick J, Hsu L-C, DeLoia J, Baysal B . Coordinate up-regulation of TMEM97 and cholesterol biosynthesis genes in normal ovarian surface epithelial cells treated with progesterone: implications for pathogenesis of ovarian cancer. BMC Cancer 2007; 7: 223.
Jira PE, Waterham HR, Wanders RJA, Smeitink JAM, Sengers RCA, Wevers RA . Smith–Lemli–Opitz syndrome and the DHCR7 gene. Ann Hum Genet 2003; 67: 269–280.
Kelley RI, RCM Hennekam . The Smith–Lemli–Opitz syndrome. J Med Genet 2000; 37: 321–335.
Prasad C, Marles S, Prasad AN, Nikkel S, Longstaffe S, Peabody D et al. Smith–Lemli–Opitz syndrome: new mutation with a mild phenotype. Am J Med Genet 2002; 108: 64–68.
Nowaczyk MJM, Irons MB . Smith–Lemli–Opitz syndrome: phenotype, natural history, and epidemiology. Am J Med Genet C: Sem Med Genet 2012; 160C: 250–262.
Fukazawa R, Nakahori Y, Kogo T, Kawakami T, Akamatsu H, Tanae A et al. Normal Y sequences in Smith–Lemli–Opitz syndrome with total failure of masculinization. Acta Paediatr 1992; 81: 570–572.
Lanthaler B, Steichen-Gersdorf E, Kollerits B, Zschocke J, Witsch-Baumgartner M . Maternal ABCA1 genotype is associated with severity of Smith–Lemli–Opitz syndrome and with viability of patients homozygous for null mutations. Eur J Hum Genet 2013; 21: 286–293.
De Brasi D, Esposito T, Rossi M, Parenti G, Sperandeo M, Zuppaldi A et al. Smith–Lemli–Opitz syndrome: evidence of T93M as a common mutation of delta7-sterol reductase in Italy and report of three novel mutations. Eur J Hum Genet 1999; 7: 937–940.
Nowaczyk MJM, Waye JS, Douketis JD . DHCR7 mutation carrier rates and prevalence of the RSH/Smith–Lemli–Opitz syndrome: where are the patients? Am J Med Genet A 2006; 140A: 2057–2062.
Waye JS, Eng B, Potter MA, Nowaczyk MJ, McFadden D, Langlois S . De novo mutation of the DHCR7 gene in a fetus with severe Smith–Lemli–Opitz (or RSH) syndrome. Am J Med Genet A 2007; 143: 1799–1801.
Witsch-Baumgartner M, Sawyer H, Haas D . Clinical utility gene card for: Smith-Lemli-Opitz Syndrome [SLOS]. Eur J Hum Genet 2013; 21.
Megías-Vericat J, Rojas L, Herrero M, Bosó V, Montesinos P, Moscardó F et al. Influence of ABCB1 polymorphisms upon the effectiveness of standard treatment for acute myeloid leukemia: a systematic review and meta-analysis of observational studies. Pharmacogenomics J 2015; 15: 109–118.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN . The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014; 133: 1–9.
HGMD. Human Gene Mutation Database 2015, Available from: http://www.hgmd.cf.ac.uk/ac/index.php. Accessed on May 2015.
Waterham HR, RCM Hennekam . Mutational spectrum of Smith–Lemli–Opitz syndrome. Am J Med Genet C: Sem Med Genet 2012; 160C: 263–284.
Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T et al. Analysis of protein-coding genetic variation in 60,706 humans. BioRxiv 2015: 030338.
Yu H, Patel SB . Recent insights into the Smith–Lemli–Opitz syndrome. Clin Genet 2005; 68: 383–391.
Oh M-Y, Kim JS, Kim JH, Cho JH, Lee BH, Kim G-H et al. A case of Smith–Lemli–Opitz syndrome confirmed by molecular analysis: review of mutation spectrum of the DHCR7 gene in Korea. J Genet Med 2014; 11: 106–110.
Nowaczyk MJM, Nakamura LM, Eng B, Porter FD, Waye JS . Frequency and ethnic distribution of the common DHCR7 mutation in Smith–Lemli–Opitz syndrome. Am J Med Genet 2001; 102: 383–386.
Kuan V, Martineau A, Griffiths C, Hypponen E, Walton R . DHCR7 mutations linked to higher vitamin D status allowed early human migration to Northern latitudes. BMC Evolut Biol 2013; 13: 144.
Waye JS, Krakowiak PA, Wassif CA, Sterner AL, Eng B, Nakamura LM et al. Identification of nine novel DHCR7 missense mutations in patients with Smith–Lemli–Opitz syndrome (SLOS). Hum Mutat 2005; 26: 59–59.
Patrono C, Dionisi-Vici C, Giannotti A, Bembi B, Digilio M, Rizzo C et al. Two novel mutations of the human Δ7-sterol reductase (DHCR7) gene in children with Smith–Lemli–Opitz syndrome. Mol Cell Probes 2002; 16: 315–318.
Nowaczyk MJM, Zeesman S, Waye JS, Douketis JD . Incidence of Smith–Lemli–Opitz syndrome in Canada: results of three-year population surveillance. J Pediatr 2004; 145: 530–535.
Porter FD . Smith–Lemli–Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 2008; 16: 535–541.
Waye J, Nakamura L, Eng B, Hunnisett L, Chitayat D, Costa T et al. Smith–Lemli–Opitz syndrome: carrier frequency and spectrum of DHCR7 mutations in Canada. J Med Genet 2002; 39: e31–e31.
Cross J, Iben J, Simpson C, Thurm A, Swedo S, Tierney E et al. Determination of the allelic frequency in Smith–Lemli–Opitz syndrome by analysis of massively parallel sequencing data sets. Clin Genet 2014; 87: 570–575.
Opitz JM, Gilbert-Barness E, Ackerman J, Lowichik A . Cholesterol and development: the RSH (‘Smith–Lemli–Opitz’) syndrome and related conditions. Fetal Pediatr Pathol 2002; 21: 153–181.
Fricke-Galindo I, Cespedes-Garro C, Rodrigues-Soares F, Naranjo MEG, Delgado A, de Andres F et al. Interethnic variation of CYP2C19 alleles, predicted phenotypes and measured metabolic phenotypes across world populations. Pharmacogenomics J 2016; 16: 113–123.
Nowaczyk MJM, Martin-Garcia D, Aquino-Perna A, Rodriguez-Vazquez M, McCaughey D, Eng B et al. Founder effect for the T93M DHCR7 mutation in Smith–Lemli–Opitz syndrome. Am J Med Genet A 2004; 125A: 173–176.
Witsch-Baumgartner M, Clayton P, Clusellas N, Haas D, Kelley R, Krajewska-Walasek M et al. Identification of 14 novel mutations in DHCR7 causing the Smith–Lemli–Opitz syndrome and delineation of the DHCR7 mutational spectra in Spain and Italy. Hum Mutat 2005; 25: 412–412.
Witsch-Baumgartner M, Ciara E, Löffler J, Menzel H, Seedorf U, Burn J et al. Frequency gradients of DHCR7 mutations in patients with Smith–Lemli–Opitz syndrome in Europe: evidence for different origins of common mutations. Eur J Hum Genet 2001; 9: 45–50.
Kozák L, Francová H, Hrabincová E, Procházková D, Jüttnerová V, Bzdúch V et al. Smith–Lemli–Opitz syndrome: molecular-genetic analysis of ten families. J Inherit Metab Dis 2000; 23: 409–412.
Cardoso M, Balreira A, Martins E, Nunes L, Cabral A, Marques M et al. Molecular studies in Portuguese patients with Smith–Lemli–Opitz syndrome and report of three new mutations in DHCR7. Mol Genet Metab 2005; 85: 228–235.
Waterham HR, Wanders RJA . Biochemical and genetic aspects of 7-dehydrocholesterol reductase and Smith–Lemli–Opitz syndrome. Biochim Biophys Acta 2000; 1529: 340–356.
Nezarati MM, Loeffler J, Yoon G, MacLaren L, Fung E, Snyder F et al. Novel mutation in the Δ-sterol reductase gene in three Lebanese sibs with Smith–Lemli–Opitz (RSH) syndrome. Am J Med Genet 2002; 110: 103–108.
Al-Owain M, Imtiaz F, Shuaib T, Edrees A, Al-Amoudi M, Sakati N et al. Smith–Lemli–Opitz syndrome among Arabs. Clin Genet 2012; 82: 165–172.
Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet 2010; 376: 180–188.
Wehr E, Trummer O, Giuliani A, Gruber H-J, Pieber TR, Obermayer-Pietsch B . Vitamin D-associated polymorphisms are related to insulin resistance and vitamin D deficiency in polycystic ovary syndrome. Eur J Endocrinol 2011; 164: 741–749.
Karras S, Anagnostis P, Naughton D, Annweiler C, Petroczi A, Goulis D . Vitamin D during pregnancy: why observational studies suggest deficiency and interventional studies show no improvement in clinical outcomes? A narrative review. J Endocrinol Investig 2015; 38: 1265–1275.
Ozkan S, Jindal S, Greenseid K, Shu J, Zeitlian G, Hickmon C et al. Replete vitamin D stores predict reproductive success following in vitro fertilization. Fert Steril 2010; 94: 1314–1319.
Lerchbaum E, Obermayer-Pietsch B . Mechanisms in endocrinology: vitamin D and fertility: a systematic review. Eur J Endocrinol 2012; 166: 765–778.
Deeb KK, Trump DL, Johnson CS . Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 2007; 7: 684–700.
Webb AR, Kline L, Holick MF . Influence of season and latitude on the cutaneous synthesis of vitamin d3: exposure to winter sunlight in boston and edmonton will not promote vitamin d3 synthesis in human skin. J Clin Endocrinol Metab 1988; 67: 373–378.
Norman AW . Sunlight, season, skin pigmentation, vitamin D, and 25-hydroxyvitamin D: integral components of the vitamin D endocrine system. Am J Clin Nutr 1998; 67: 1108–1110.
Moebius FF, Fitzky BU, Lee JN, Paik YK, Glossmann H . Molecular cloning and expression of the human delta7-sterol reductase. Proc Natl Acad Sci USA 1998; 95: 1899–1902.
Zou L, Porter TD . Rapid suppression of 7-dehydrocholesterol reductase activity in keratinocytes by vitamin D. J Steroid Biochem Mol Biol 148: 64–71.
Rossi M, Federico G, Corso G, Parenti G, Battagliese A, Frascogna AR et al. Vitamin D status in patients affected by Smith–Lemli–Opitz syndrome. J Inherit Metab Dis 2005; 28: 69–80.
Hill AB . The environment and disease: association or causation? Proc R Soc Med 1965; 58: 295.
Lauth M, Rohnalter V, Bergstrom A, Kooshesh M, Svenningsson P, Toftgard R . Antipsychotic drugs regulate hedgehog signaling by modulation of 7-dehydrocholesterol reductase levels. Mol Pharmacol 2010; 78: 486–496.
Fernø J, Raeder M, Vik-Mo A, Skrede S, Glambek M, Tronstad K et al. Antipsychotic drugs activate SREBP-regulated expression of lipid biosynthetic genes in cultured human glioma cells: a novel mechanism of action? Pharmacogenomics J 2005; 5: 298–304.
Raeder MB, Fernø J, Vik-Mo AO, Steen VM . SREBP activation by antipsychotic-and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol Cell Biochem 2006; 289: 167–173.
Hall P, Michels V, Gavrilov D, Matern D, Oglesbee D, Raymond K et al. Aripiprazole and trazodone cause elevations of 7-dehydrocholesterol in the absence of Smith–Lemli–Opitz syndrome. Mol Genet Metab 2013; 110: 176–178.
Rodgers MA, Villareal VA, Schaefer EA, Peng LF, Corey KE, Chung RT et al. Lipid metabolite profiling identifies desmosterol metabolism as a new antiviral target for hepatitis C virus. J Am Chem Soc 2012; 134: 6896–6899.
Davis AP, Grondin CJ, Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL et al. The Comparative Toxicogenomics Database's 10th year anniversary: update 2015. Nucleic Acids Res 2015; 43: D914–D920.
CTD.Comparative Toxicogenomics Database. Available from: http://ctdbase.org/. Accessed on February 2015.
Foley D, Mackinnon A . A systematic review of antipsychotic drug effects on human gene expression related to risk factors for cardiovascular disease. Pharmacogenomics J 2014; 14: 446–451.
Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014; 42: D1083–D1090.
Keller TH, Pichota A, Yin Z . A practical view of ‘druggability’. Curr Opin Chem Biol 2006; 10: 357–361.
Hopkins AL, Groom CR . The druggable genome. Nat Rev Drug Discov 2002; 1: 727–730.
Teo CC, Kon OL, Sim KY, Ng SC . Synthesis of 2-(p-chlorobenzyl)-3-aryl-6-methoxybenzofurans as selective ligands for antiestrogen-binding sites. Effects on cell proliferation and cholesterol synthesis. J Med Chem 1992; 35: 1330–1339.
E.O.R.T.C. Breast Cancer Group. Clinical trial of nafoxidine, an oestrogen antagonist in advanced breast cancer. Eur J Cancer 1972; 8: 387–389.
Engelsman E, Heuson JC, Blonk, Van Der Wijst J, Drochmans A, Maass H et al. Controlled clinical trial of l-dopa and nafoxidine in advanced breast cancer: an E.O.R.T.C. study. Br Med J 1975; 2: 714–715.
Jain J, Samal B, Singhakowinta A, Vaitkevicius VK . Clinical trial of nafoxidine in adrenalectomized patients with advanced breast cancer. Cancer 1977; 40: 2063–2066.
Steinbaum FL, De Jager RL, Krakoff IH . Clinical trial of nafoxidine in advanced breast cancer. Med Pediat Oncol 1978; 4: 123–126.
Finnell RH . Teratology: general considerations and principles. J Allergy Clin Immunol 1999; 103: S337–S342.
Beckman D, Brent R . Mechanisms of teratogenesis. Ann Pharmacol Toxicol 1984; 24: 483–500.
Roach E, Demyer W, Conneally P, Palmer C, Merritt A . Holoprosencephaly: birth data, genetic and demographic analyses of 30 families. Birth Defects Orig Artic Ser 1974; 11: 294–313.
Shim Y-H, Bae S-H, Kim J-H, Kim K-R, Kim CJ, Paik Y-K . A novel mutation of the human 7-dehydrocholesterol reductase gene reduces enzyme activity in patients with holoprosencephaly. Biochem Biophys Res Commun 2004; 315: 219–223.
Edison RJ, Muenke M . Gestational exposure to lovastatin followed by cardiac malformation misclassified as holoprosencephaly. N Engl J Med 2005; 352: 2759–2759.
Edison RJ, Muenke M . Central nervous system and limb anomalies in case reports of first-trimester statin exposure. N Engl J Med 2004; 350: 1579–1582.
Ventura SJ, Curtin SC, Abma JC, Henshaw SK . Estimated pregnancy rates and rates of pregnancy outcomes for the United States, 1990–2008. Natl Vital Stat Rep 2012; 60: 1–21.
Control CfD, Prevention. Update on overall prevalence of major birth defects—Atlanta, Georgia, 1978–2005. MMWR Morbid Mortal Week Rep 2008; 57: 1–5.
Germann N, Goffinet F, Goldwasser F . Anthracyclines during pregnancy: embryo–fetal outcome in 160 patients. Ann Oncol 2004; 15: 146–150.
Tewari K, Bonebrake RG, Asrat T, Shanberg AM . Ambiguous genitalia in infant exposed to tamoxifen in utero. Lancet 1997; 350: 183.
Barthelmes L, Gateley C . Tamoxifen and pregnancy. Breast 2004; 13: 446–451.
Braems G, Denys H, De Wever O, Cocquyt V, Van den Broecke R . Use of tamoxifen before and during pregnancy. Oncologist 2011; 16: 1547–1551.
Berger JC, Clericuzio CL . Pierre Robin sequence associated with first trimester fetal tamoxifen exposure. Am J Med Genet A 2008; 146: 2141–2144.
Cullins SL, Pridjian G, Sutherland CM . Goldenhar's syndrome associated with tamoxifen given to the mother during gestation. JAMA 1994; 271: 1905–1906.
Murray CL, Reichert JA, Anderson J, Twiggs LB . Multimodal cancer therapy for breast cancer in the first trimester of pregnancy: a case report. Jama 1984; 252: 2607–2608.
Potluri V, Lewis D, Burton GV . Chemotherapy with taxanes in breast cancer during pregnancy: case report and review of the literature. Clin Breast Cancer 2006; 7: 167–170.
Roth DE . Vitamin D supplementation during pregnancy: safety considerations in the design and interpretation of clinical trials. J Perinatol 2011; 31: 449–459.
De-Regil LM, Palacios C, Ansary A, Kulier R, Pena-Rosas JP . Vitamin D supplementation for women during pregnancy. Cochrane Database Syst Rev 2012; 2: CD008873.
De-Regil LM, Palacios C, Lombardo LK, Peña-Rosas JP . Vitamin D supplementation for women during pregnancy. Cochrane Database of Syst Rev 2016; 14: CD008873.
Miliku K, Voortman T, Franco OH, McGrath JJ, Eyles DW, Burne TH et al. Vitamin D status during fetal life and childhood kidney outcomes. Eur J Clin Nutr 2015; 70: 629–634.
Correa-Cerro LS, Wassif CA, Waye JS, Krakowiak PA, Cozma D, Dobson NR et al. DHCR7 nonsense mutations and characterisation of mRNA nonsense mediated decay in Smith–Lemli–Opitz syndrome. J Med Genet 2005; 42: 350–357.
Ahn J, Yu K, Stolzenberg-Solomon R, Simon KC, McCullough ML, Gallicchio L et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet 2010; 19: 2739–2745.
Jira P, Wanders R, Smeitink J, De Jong J, Wevers R, Oostheim W et al. Novel mutations in the 7-dehydrocholesterol reductase gene of 13 patients with Smith–Lemli–Opitz syndrome. Ann Hum Genet 2001; 65: 229–236.
Evans T, Poh A, Webb C, Wainwright B, Wicking C, Glass I et al. Novel mutation in the Δ7-dehydrocholesterol reductase gene in an Australian patient with Smith-Lemli-Opitz syndrome. Am J Med Genet 2001; 103: 344–347.
Goldenberg A, Chevy F, Bernard C, Wolf C, Cormier-Daire V . Circonstances cliniques du diagnostic du syndrome de Smith-Lemli-Opitz et tentatives de corrélation phénotype-génotype: à propos de 45 cas. Arch Pédiatrie 2003; 10: 4–10.
Scalco F, Correa-Cerro L, Wassif C, Porter F, Moretti-Ferreira D . DHCR7 mutations in Brazilian Smith–Lemli–Opitz syndrome patients. Am J Med Genet A 2005; 136: 278–281.
Ginat S, Battaile KP, Battaile BC, Maslen C, Gibson KM, Steiner RD . Lowered DHCR7 activity measured by ergosterol conversion in multiple cell types in Smith–Lemli–Opitz syndrome. Mol Genet Metab 2004; 83: 175–183.
Jezela-Stanek A, Ciara E, Małunowicz E, Chrzanowska K, Latos-Bieleńska A, Krajewska-Walasek M et al. Differences between predicted and established diagnoses of Smith-Lemli-Opitz syndrome in the Polish population: underdiagnosis or loss of affected fetuses? J Inherit Metab Dis 2010; 33: 241–248.
Matsumoto Y, Morishima K-i, Honda A, Watabe S, Yamamoto M, Hara M et al. R352Q mutation of the DHCR7 gene is common among Japanese Smith–Lemli–Opitz syndrome patients. J Hum Genet 2005; 50: 353–356.
Anstey AV, Azurdia RM, Rhodes LE, Pearse AD, Bowden PE . Photosensitive Smith–Lemli–Opitz syndrome is not caused by a single gene mutation: analysis of the gene encoding 7-dehydrocholesterol reductase in five UK families. Br J Dermatol 2005; 153: 774–779.
Waterham HR, Wijburg FA, Hennekam RCM, Vreken P, Poll-The BT, Dorland L et al. Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet 1998; 63: 329–338.
Szabó G, Oláh A, Kozak L, Balogh E, Nagy A, Blahakova I et al. A patient with Smith–Lemli–Opitz syndrome: novel mutation of the DHCR7 gene and effects of therapy with simvastatin and cholesterol supplement. Eur J Pediatr 2010; 169: 121–123.
Romano F, Fiore B, Pezzino FM, Longombardo MT, Cefalù AB, Noto D et al. A novel mutation of the DHCR7 gene in a Sicilian compound heterozygote with Smith–Lemli–Opitz syndrome. Mol Diagn 2005; 9: 201–204.
Ferno J, Skrede S, Vik-Mo A, Havik B, Steen V . Drug-induced activation of SREBP-controlled lipogenic gene expression in CNS-related cell lines: marked differences between various antipsychotic drugs. BMC Neurosci 2006; 7: 69.
Wassif CA, Krakowiak PA, Wright BS, Gewandter JS, Sterner AL, Javitt N et al. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith–Lemli–Opitz syndrome fibroblasts. Mol Genet Metab 2005; 85: 96–107.
Correa-Cerro LS, Wassif CA, Kratz L, Miller GF, Munasinghe JP, Grinberg A et al. Development and characterization of a hypomorphic Smith–Lemli–Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet 2006; 15: 839–851.
Park E-J, Zahari NEM, Lee E-W, Song J, Lee J-H, Cho M-H et al. SWCNTs induced autophagic cell death in human bronchial epithelial cells. Toxicol In Vitro 2014; 28: 442–450.
Guo L, Xiao Y, Wang Y . Hexavalent chromium-induced alteration of proteomic landscape in human skin fibroblast cells. J Proteome Res 2013; 12: 3511–3518.
Kawata K, Yokoo H, Shimazaki R, Okabe S . Classification of heavy-metal toxicity by human DNA microarray analysis. Environ Sci Technol 2007; 41: 3769–3774.
Hundhausen C, Boesch-Saadatmandi C, Matzner N, Lang F, Blank R, Wolffram S et al. Ochratoxin A Lowers mRNA levels of genes encoding for key proteins of liver cell metabolism. Cancer Genomics Proteomics 2008; 5: 319–332.
Guo L, Xiao Y, Wang Y . Monomethylarsonous acid inhibited endogenous cholesterol biosynthesis in human skin fibroblasts. Toxicol Appl Pharmacol 2014; 277: 21–29.
Ruenitz PC, Arrendale RF, Schmidt WF, Thompson CB, Nanavati NT . Phenolic metabolites of clomiphene: [(E,Z)-2-[4-(1,2-diphenyl-2-chlorovinyl)phenoxy]ethyl]diethylamine. Preparation, electrophilicity, and effects in MCF 7 breast cancer cells. J Med Chem 1989; 32: 192–197.
Jordan VC . Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J Med Chem 2003; 46: 883–908.
Bignon E, Pons M, Crastes de Paulet AC, Dore JC, Gilbert J, Abecassis J et al. Effect of triphenylacrylonitrile derivatives on estradiol-receptor binding and on human breast cancer cell growth. J Med Chem 1989; 32: 2092–2103.
Burke PJ, Kalet BT, Koch TH . Antiestrogen binding site and estrogen receptor mediate uptake and distribution of 4-hydroxytamoxifen-targeted doxorubicin-formaldehyde conjugate in breast cancer cells. J Med Chem 2004; 47: 6509–6518.
Chang LC, Bhat KP, Pisha E, Kennelly EJ, Fong HH, Pezzuto JM et al. Activity-guided isolation of steroidal alkaloid antiestrogen-binding site inhibitors from Pachysandra procumbens. J Nat Prod 1998; 61: 1257–1262.
Sharma AP, Saeed A, Durani S, Kapil RS . Structure-activity relationship of antiestrogens. Phenolic analogues of 2,3-diaryl-2H-1-benzopyrans. J Med Chem 1990; 33: 3222–3229.
Taylor PN, Minassian C, Rehman A, Iqbal A, Draman MS, Hamilton W et al. TSH levels and risk of miscarriage in women on long-term levothyroxine: a community-based study. J Clin Endocrinol Metab 2014; 99: 3895–3902.
Neto LV, De Almeida CA, Da Costa SM, Vaisman M . Prospective evaluation of pregnant women with hypothyroidism: Implications for treatment. Gynecol Endocrinol 2007; 23: 138–141.
Zamperini P, Gibelli B, Gilardi D, Tradati N, Chiesa F . Pregnancy and thyroid cancer: ultrasound study of foetal thyroid. Acta Otorhinolaryngol Ital 2009; 29: 339.
Pomorski L, Bartos M, Narebski J . Pregnancy following operative and complementary treatment of thyroid cancer. Zentralbl Gynakol 1999; 122: 383–386.
Rotondi M, Caccavale C, Di Serio C, Del Buono A, Sorvillo F, Glinoer D et al. Successful outcome of pregnancy in a thyroidectomized-parathyroidectomized young woman affected by severe hypothyroidism. Thyroid 1999; 9: 1037–1040.
Gentile S . Clinical utilization of atypical antipsychotics in pregnancy and lactation. Ann Pharmacother 2004; 38: 1265–1271.
McKenna K, Koren G, Tetelbaum M, Wilton L, Shakir S, Diav-Citrin O et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry 2005; 66: 444–449, quiz 546.
Karakuła H, Szajer K, Rpila B, Grzywa A, Chuchra M . Clozapine and pregnancy—a case history. Pharmacopsychiatry 2004; 37: 303–304.
Stoner SC, Sommi R, Marken PA, Anya I, Vaughn J . Clozapine use in two full-term pregnancies. J Clin Psychiatry 1997; 58: 364–365.
Reis M, Källén B . Maternal use of antipsychotics in early pregnancy and delivery outcome. J Clin Psychopharmacol 2008; 28: 279–288.
Ahn H, Choi J, Han J, Kim M, Chung J, Ryu H et al. Pregnancy outcome after exposure to oral contraceptives during the periconceptional period. Hum Exp Toxicol 2008; 27: 307–313.
Aarskog D . Maternal progestins as a possible cause of hypospadias. N Engl J Med 1979; 300: 75–78.
Hayles AB, Nolan RB . Masculinization of female fetus, possibly related to administration of progesterone during pregnancy; report of two cases. Proc Staff Meet Mayo Clin 1958; 33: 200–203.
Mendhekar D, Andrade C . Uneventful use of haloperidol and trihehexyphenidyl during three consecutive pregnancies. Arch Women's Ment Health 2011; 14: 83–84.
Diav-Citrin O, Shechtman S, Ornoy S, Arnon J, Schaefer C, Garbis H et al. Safety of haloperidol and penfluridol in pregnancy: a multicenter, prospective, controlled study. J Clin Psychiatry 2005; 66: 317–322.
Yaris F, Yaris E, Kadioglu M, Ulku C, Kesim M, Kalyoncu NI . Use of polypharmacotherapy in pregnancy: a prospective outcome in a case. Prog Neuro-Psychopharmacol Biol Psychiatry 2004; 28: 603–605.
Newport DJ, Calamaras MR, DeVane CL, Donovan J, Beach AJ, Winn S et al. Atypical antipsychotic administration during late pregnancy: placental passage and obstetrical outcomes. Am J Psychiatry 2007; 164: 1214–1220.
Sawhney H, Vasishta K, Rani K . Comparison of lytic cocktail and magnesium sulphate regimens in eclampsia: a retrospective analysis. J Obstet Gynaecol Res 1998; 24: 261–266.
Chatterjee A, Mukheree J . Comparative study of different anticonvulsants in eclampsia. J Obstet Gynaecol Res 1997; 23: 289–293.
Taguchi N, Rubin ET, Hosokawa A, Choi J, Ying AY, Moretti ME et al. Prenatal exposure to HMG-CoA reductase inhibitors: effects on fetal and neonatal outcomes. Reprod Toxicol 2008; 26: 175–177.
Pollack PS, Shields KE, Burnett DM, Osborne MJ, Cunningham ML, Stepanavage ME . Pregnancy outcomes after maternal exposure to simvastatin and lovastatin. Birth Defects Res A Clin Mol Teratol 2005; 73: 888–896.
Edison RJ, Muenke M . Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet A 2004; 131: 287–298.
Manson JM, Freyssinges C, Ducrocq MB, Stephenson WP . Postmarketing surveillance of lovastatin and simvastatin exposure during pregnancy. Reprod Toxicol 1996; 10: 439–446.
Andreadis C, Charalampidou M, Diamantopoulos N, Chouchos N, Mouratidou D . Combined chemotherapy and radiotherapy during conception and first two trimesters of gestation in a woman with metastatic breast cancer. Gynecol Oncol 2004; 95: 252–255.
Öksüzoglu B, Güler N . An infertile patient with breast cancer who delivered a healthy child under adjuvant tamoxifen therapy. Eur J Obstet Gynecol Reprod Biol 2002; 104: 79.
Koizumi K, Aono T . Pregnancy after combined treatment with bromocriptine and tamoxifen in two patients with pituitary prolactinomas. Fertil Steril 1986; 46: 312–314.
Isaacs R, Hunter W, Clark K . Tamoxifen as systemic treatment of advanced breast cancer during pregnancy—case report and literature review. Gynecol Oncol 2001; 80: 405–408.
Clark S . Prophylactic tamoxifen. Lancet 1993; 342: 168.
Cardonick E, Gilmandyar D, Somer RA . Maternal and neonatal outcomes of dose-dense chemotherapy for breast cancer in pregnancy. Obstet Gynecol 2012; 120: 1267–1272.
Mir O, Berrada N, Domont J, Cioffi A, Boulet B, Terrier P et al. Doxorubicin and ifosfamide for high-grade sarcoma during pregnancy. Cancer Chemother Pharmacol 2012; 69: 357–367.
Morris PG, King F, Kennedy MJ . Cytotoxic chemotherapy for pregnancy-associated breast cancer: single institution case series. J Oncol Pharm Pract 2009; 15: 241–247.
de Wildt SN, Taguchi N, Koren G . Unintended pregnancy during radiotherapy for cancer. Nat Clin Pract Oncol 2009; 6: 175–178.
Soliman KB, Abbas MM, Seksaka MA, Wafa S, Balah AS . Aggressive primary thyroid non Hodgkin's lymphoma with pregnancy. Saudi Med J 2007; 28: 634–636.
Hahn KM, Johnson PH, Gordon N, Kuerer H, Middleton L, Ramirez M et al. Treatment of pregnant breast cancer patients and outcomes of children exposed to chemotherapy in utero. Cancer 2006; 107: 1219–1226.
Nieto Y, Santisteban M, Aramendía JM, Fernández-Hidalgo Ó, García-Manero M, López G . Docetaxel administered during pregnancy for inflammatory breast carcinoma. Clin Breast Cancer 2006; 6: 533–534.
Peccatori F, Martinelli G, Gentilini O, Goldhirsch A . Chemotherapy during pregnancy: what is really safe? Lancet Oncol 2004; 5: 398.
Willemse P, Van der Sijde R, Sleijfer DT . Combination chemotherapy and radiation for stage IV breast cancer during pregnancy. Gynecol Oncol 1990; 36: 281–284.
Karp G, Von Oeyen P, Valone F, Khetarpal V, Israel M, Mayer R et al. Doxorubicin in pregnancy: possible transplacental passage. Cancer Treat Rep 1983; 67: 773–777.
Kerr JR . Neonatal effects of breast cancer chemotherapy administered during pregnancy. Pharmacotherapy 2005; 25: 438–441.
Meyer-Wittkopf M, Barth H, Emons G, Schmidt S . Fetal cardiac effects of doxorubicin therapy for carcinoma of the breast during pregnancy: case report and review of the literature. Ultrasound Obstet Gynecol 2001; 18: 62–66.
Barni S, Ardizzoia A, Zanetta G, Strocchi E, Lissoni P, Tancini G . Weekly doxorubicin chemotherapy for breast cancer in pregnancy. A case report. Tumori 1992; 78: 349–350.
Schaefer C, Meister R, Weber-Schoendorfer C . Isotretinoin exposure and pregnancy outcome: an observational study of the Berlin Institute for Clinical Teratology and Drug Risk Assessment in Pregnancy. Arch Gynecol Obstet 2010; 281: 221–227.
Honein M, Paulozzi L, Erickson J . Continued occurrence of Accutane-exposed pregnancies. Teratology 2001; 64: 142–147.
Dai WS, LaBraico JM, Stern RS . Epidemiology of isotretinoin exposure during pregnancy. J Am Acad Dermatol 1992; 26: 599–606.
Acknowledgements
MRB supported by the National Library of Medicine training grant T15 LM00707, and R01 GM107145 supported this work.
Author contributions
Mary Regina Boland performed literature review and analyses, synthesized literature and wrote the manuscript. Nicholas P Tatonetti provided feedback on the review findings, provided insights regarding the cohesiveness of the review, and edited the text of the manuscript in addition to providing funding support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors have no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the The Pharmacogenomics Journal website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Boland, M., Tatonetti, N. Investigation of 7-dehydrocholesterol reductase pathway to elucidate off-target prenatal effects of pharmaceuticals: a systematic review. Pharmacogenomics J 16, 411–429 (2016). https://doi.org/10.1038/tpj.2016.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2016.48
This article is cited by
-
Medication effects on developmental sterol biosynthesis
Molecular Psychiatry (2022)
-
Trazodone effects on developing brain
Translational Psychiatry (2021)
-
Immunopathobiology and therapeutic targets related to cytokines in liver diseases
Cellular & Molecular Immunology (2021)
-
Autism Spectrum Disorder: Signaling Pathways and Prospective Therapeutic Targets
Cellular and Molecular Neurobiology (2021)
-
Enabling pregnant women and their physicians to make informed medication decisions using artificial intelligence
Journal of Pharmacokinetics and Pharmacodynamics (2020)
